Quality Control in a Virology Laboratory
In this section, various aspects of quality control in a virology laboratory will be looked at. The following topics will be covered:
General Principles of Quality Control
The validity of diagnostic test results produced in each laboratory is entirely dependent on the measures employed before, during, and after each assay. Consistency in the production of good results requires an overall program that includes quality assurance, quality control, and quality assessment.
Definitions
Quality Control - QC refers to the measures that must be included during each assay to verify that the test is working properly.
Quality Assurance - QA is defined as the overall program that ensures that the final results reported by the laboratory are correct
Quality Assessment - quality assessment (also known as proficiency testing) is a means to determine the quality of the results generated by the laboratory. It is usually an external evaluation of the laboratory's performance. Internal quality assessment programs can also be instituted. Quality assessment is a challenge to the effectiveness of the QA and QC programs.
"The aim of quality control is simply to ensure that the results generated by the test are correct. However, quality assurance is concerned with much more: that the right test is carried out on the right specimen, and that the right result and right interpretation is delivered to the right person at the right time"
Many variables can affect the quality of results
Quality Assurance
QA is an ongoing process that requires daily attention by all laboratory staff. Some fundamental issues in QA related to specimens include;
Inspection of all specimens upon receipt and before testing to ensure that they are suitable, Lipaemic, haemolysed or contaminated samples should not be used as these may interfere with assay performance.
QA also includes such factors as;
The following sections indicate components that must be continually monitored and represent fundamental aspects of a good QA program
A. Record Keeping
An efficient laboratory will be able to monitor the records of specimens from the time the samples arrive until the time that results are released. Logbooks are an essential step in the recording of laboratory specimens and should be kept confidential. Any specimen that is determined to be inadequate for testing or that does not contain the essential information e.g. for HIV testing, should not be tested and a note should be entered in the logbook. A worksheet must accompany each test run in the laboratory. The worksheet serves as a guide when placing samples on the run. QC records are important in validating laboratory results. A standard operating procedure manual should be kept in the laboratory at all times and should be reviewed and updated frequently.
B. Monitoring Laboratory Staff
Laboratory managers may wish to periodically monitor the performance of their laboratory staff. Samples with known results may be resubmitted discreetly along with the routine workload.
C. Vigilance in the Laboratory
Vigilance refers to the watchfulness and consists of always
D. Verification of True Positive and Negatives
A positive HIV result is a serious concern and each laboratory must be absolutely certain that each positive specimen result is correct. Once a sample is found to be positive by a screening test, an aliquot from the initial specimen tube should be retested, Wherever possible, a second specimen should be collected from the individual and rested to eliminate any possible handling, labeling, or clerical errors.
E. Parallel Testing of Resubmitted specimens
Parallel testing of resubmitted specimens is important in those patients whose specimens have yielded indeterminate results.
F. Reviewing Transcriptional measures
Transcriptional or clerical errors include mistakes made during the transfer of information from the test readout to the worksheet, and from the worksheet to the computer or report form. These type of errors probably account for the majority of errors in the laboratory. Possible mechanisms include having a second technologist to check the final result and the supervisor to check the results before releasing.
G. Reporting of Results
In the case of HIV testing, the handling of the results must be controlled so that the confidentiality of all persons tested is protected. A policy decision on the handling of HIV test results must be established and uniformly enforced in the laboratory. When an indeterminate result is reported, care should be taken to ensure that the clinician understands the significance of such a result and the importance of a follow-up specimen.
H. Interaction with Physicians
The relationship between laboratory personnel and physicians should be one of mutual trust and respect.
I. Storage of specimens for follow-up testing
Once a specimen has been tested, it should be stored at -20oC in the appropriate vial. An organized serum or other specimen bank should be available
J. Laboratory Efficiency
The aim is to provide turnaround time for submitted samples. Certain items must be periodically evaluated to increase efficiency in the laboratory.
K. Total Quality Management
TQM refers to a comprehensive organizational approach that is focused on continually improving the quality and efficiency with which the laboratory operates. QA is a defined program that is focused on maximizing detection of laboratory error, while TQM aims to assist in this process by maximizing efficiency. TQM is not only concerned with the monitoring of the QC/QA program, but should also include other technical or administrative considerations that may indirectly influence the quality and efficiency of the laboratory operation. This includes the evaluation of the laboratory staff and continuing education.
Quality Control: Monitoring the Testing Process
As mentioned previously, QC refers to those measures that must be included during each assay in order to verify that the test is working properly. The following items are essential elements of quality control that must be performed during every assay:
Ordinarily, each test kit has a set of positive and negative control that is to be included in each test run. These controls are considered to be internal controls, while any other controls included in the run are referred to as external controls. Internal controls are essential for QC measures for each run and are intended for use only with the lot number of the corresponding test kit. External controls can be included on a run to monitor consistent performance, lot to lot variation between kits, and to serve as an indicator of assay performance on samples that are borderline reactors.
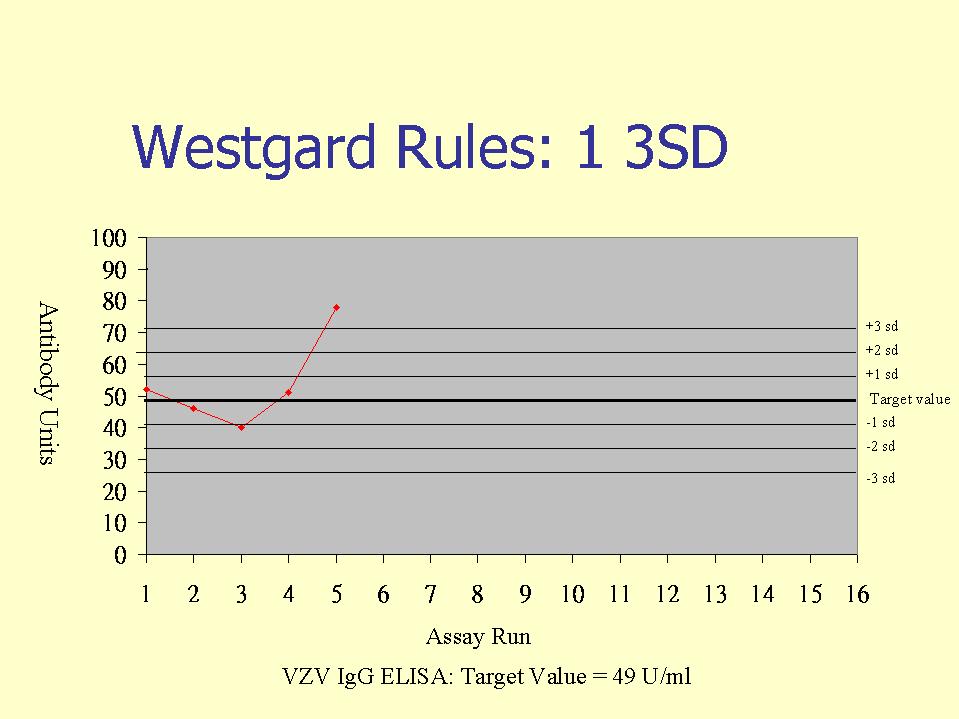
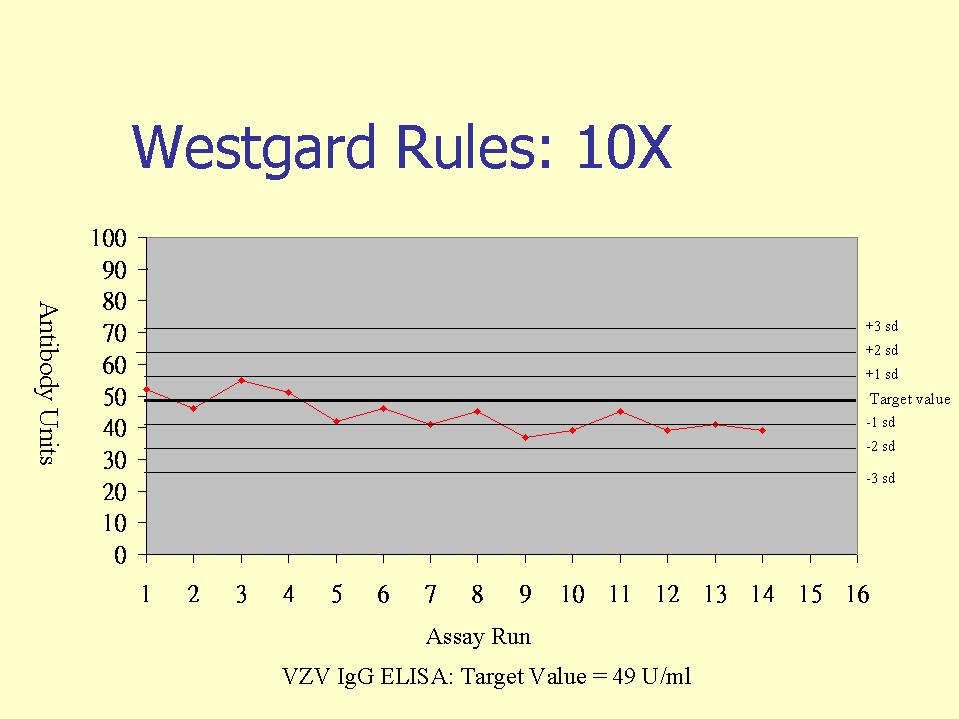
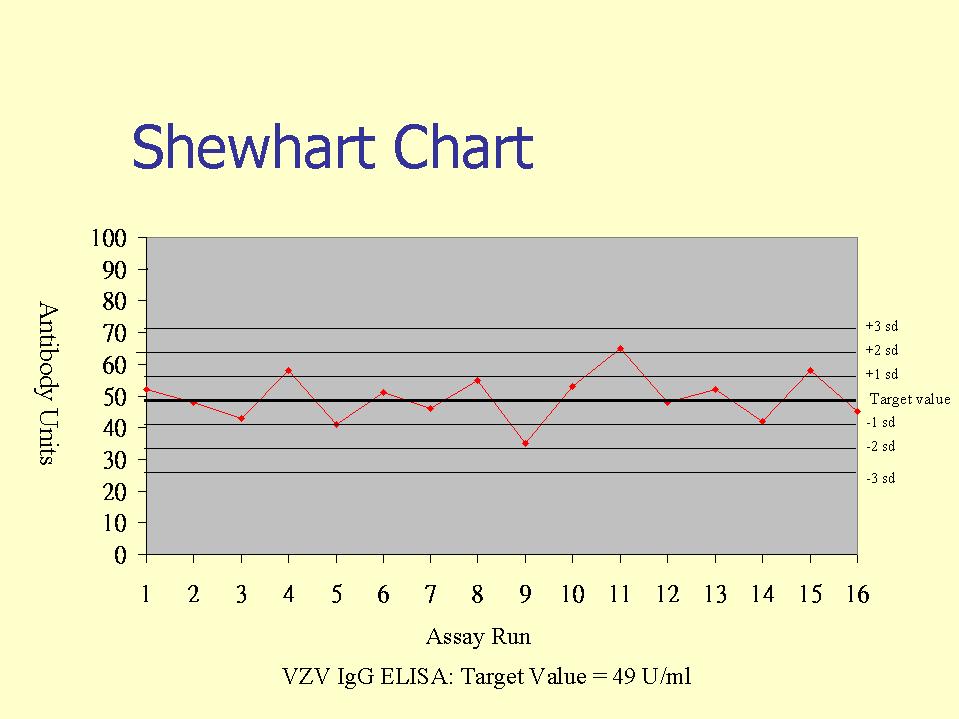
External controls, or otherwise known as internal quality control (IQC) specimens are used in internal quality control programs, whereby IQC samples are included in serological assays. The IQC samples are then evaluated against Westgard rules, whereby the IQC values are plotted in a Shewhart type chart (this may be in terms of arbitrary units or IQC o.d./Cut-off o.d.). Westgard rules define specific performance limits and are designed to detect both random and systematic errors. Of the six commonly used Westgard rules, three are warning rules and the other three are mandatory rules. The latter, if broken should result in the rejection of the test run.
Warning Rules
Four consecutive control values exceed the same limit. This again indicates the possible presence of systematic errors and may indicate the need to perform instrument maintenance or equipment calibration.
Mandatory rules
10 consecutive values are on the same side of the mean or target value. This detects systematic errors. This may happen when a new test batch or introduced or changes in the calibration of equipment.
Quality Assessment
Quality assessment is a means to determine the quality of results. It is usually an external evaluation of a laboratory's performance that relies on incorporating proficiency panels of well-characterized sera into the testing routine. External quality assessment (EQA) is now recognized as an essential component of quality assurance and is the only means to give the laboratory manager an independent means of ensuring that his routine quality control is adequate and effective. The National External Quality Assessment Scheme (NEQAS) is administered by the PHLS
The following are distributed under the NEQAS scheme:
It is important for the participating laboratory to treat NEQAS specimens in the same manner as normal routine specimens. Rubella IgM and HIV currently lack national reference preparations, as opposed to rubella IgG and HBsAg. Occasionally, more difficult specimens than usual are distributed as "education" material, such as those containing very low levels of HBsAg or anti-HIV. Scores are not normally allocated for this type of specimen, but participants may find it useful to compare their results with those of their peers.
The scoring system for NEQAS is as follows
2 fully correct report
1 partially correct report e.g. partial identification of a virus or an equivocal serological result
0 erroneous result which would not have serious clinical consequences e.g. false negative rubella IgG
-1 erroneous result which would have serious clinical consequences, e.g. false negative HBsAg or anti-HIV
It is highly desirable for the laboratory itself to have an internal quality assessment program, whereby anonymous clinical samples are submitted to the laboratory. An internal quality assessment scheme can be used to monitor the quality of the work more frequently and accurately than EQA schemes, since EQA samples are usually received infrequently and they are usually treated differently from the routine specimens. Experience at laboratories that have an internal quality assessment scheme has generally been that internal schemes are much better at identifying quality problems in the laboratory than external schemes.
Quality Control in Clinical Virology
Quality control in the clinical laboratory consists of a set of procedures designed to help ensure delivery to the medical staff of laboratory results that are consistent and accurate. These results must be supplied in a timely fashion while the data is still clinically relevant. A clinical virology laboratory should be designed in a manner so that biohazard risks to the laboratory personnel and the general public is minimized and that cultures are protected from environmental contamination. A facility designed specifically for clinical virology should;-
Be physically separate from the microbiology laboratory and not share common air returns or equipment such as hoods and incubators.
The environment should be controlled so that the ambient temperature is 22 - 26oC and the relative humidity 30 -50%.
The facility should be under negative pressure with respect to the rest of the laboratory area.
Internally, the laboratory can be divided into positive and negative air pressure areas; the positive for tissue culture and media preparation, the negative for viral isolation or serology because they deal with viable pathogens.
All surfaces should be composed of materials that can be decontaminated easily.
Good standard microbiology measures should be observed such as daily decontamination of all work surfaces, proper laboratory attire, use of safe pipetting devices, and to minimize aerosol generation.
Biological safety hoods should be available for tissue culture and viral isolation. Hood rooms should not have common air ducting and the exhaust from hoods in which pathogens are handled be externally vented.
Written Standard Operating Procedures
Two sets of written standard operating procedures are important. One is for use by medical staff and the other for laboratory use as procedure manuals. The procedures for medical staff should include;-
The procedure manual used in the laboratory should be complete enough in detail so that an inexperienced technologist can perform the procedure without additional information. One copy of the manual should be readily available to bench personnel, another copy should be stored separately in case of accidents.
Specimen Transport
Specimens for viral isolation are often held for long periods of time before it reaches the laboratory. Enveloped viruses such as RSV and CMV are extremely liable to room temperature and freeze-thaw cycles whereas non-enveloped viruses such as enteroviruses tolerate these conditions well. As a general rule, viral specimens held for short periods should be refrigerated, while those for longer periods may be frozen at -20 or -70oC.
Transport Media - the composition and type of viral transport media can affect viral isolation rates. In general, the media should be a balanced isotonic solution at physiological pH. It should contain a substance that will stabilize the virus such as gelatin, fetal calf serum or bovine serum albumin, and antibiotics against bacteria and fungi. The swab should be made of a material that is non-toxic to viruses, such as dacron or rayon.
Smears - smears are becoming increasingly popular because of the rapidity of staining techniques. The smear should contain a reasonable number of cells, be of a reasonable size and not over-contaminated by blood or pus, as the latter may lead to nonspecific staining.
Specimens for Serology - Excessively haemolysed, lipaemic, bacterially contaminated, or leaking specimens should be rejected. Sera should be heat-inactivated depending on the tests to be performed. In the event of a specimen being rejected, the ward must be informed, preferably by an oral report followed by a written one. Extenuating circumstances may warrant the acceptance of a substandard specimen.
Tissue Culture and Media
Tissue culture remain the mainstay of non-serologically viral diagnosis. Therefore, adequate quality control for commercially purchased or for in-house preparation of tissue culture cells is of great importance. Within a given cell line, there may be significant variations in sensitivity to virus isolation which may depend on the particular cell subline or clone and the passage number. Information on a particular cell line should be recorded pertinently including the source, type, passage number, confluency and cell condition. The loss of a cell line routinely passaged for use can lead to a severe disruption of workflow and therefore, provisions must be made for back-up cells in the event of contamination or laboratory accident. These back-up systems include;
Cells purchased from a commercial company should be certified to be free from mycoplasma, fungal, and bacterial contamination and examined for contamination on receipt. Tissue culture lines carried in-house should be subjected to;
Other quality control procedures that may aid in minimizing the risk of contamination include the exclusion of laboratory with infectious diseases from handling tissue culture, separate laboratory apparel, reagents and glassware for tissue culture. Cell lines should be handled separately and the cabinet be decontaminated in between.
Media - following filter sterilization, aliquots of the media should be taken and checked for bacteriological or fungal investigation. These samples should be examined daily for 5 days and should be free from contamination before that lot of medium is released for use. Aliquots of all other medium components such as fetal calf serum and L-glutamine should also be checked. New lots of medium and fetal calf serum that have passed the sterility check should be monitored for their ability to support cell growth.
Reagents and Kits
Reagents and kits should be ordered from reputable manufacturers or dealers with reliable transportation systems. Upon receipt, the reagents should be checked for obvious breakage or contamination. The quantity, source, lot number and date of receipt should be entered in a logbook and the reagents stored according to the manufacturer’s storage specifications. When new lots of any reagent are opened, the date should be noted on the container. Caution must be exercised in the case of kits as different components of a kit may have different storage conditions and expiration dates.
Instruments
Laboratory instruments should be subjected to routine preventive maintenance and checked and calibrated on a regular basis. Some of these checks can be performed by laboratory staff and entered into a logbook. The following are some recommendations for routine laboratory maintenance and performance checks on instruments.
Safety cabinets - daily air pressure check and cleaning of UV lamp. Work surface should be decontaminated after each use. Annual checks for air velocity and filter integrity and paraldehyde decontamination as applicable.
Pathology Department Accreditation
In the past two decades, there is a movement towards the accreditation of medical testing laboratories worldwide. Accreditation is an external audit of an applicant department’s organization and quality assurance program. There is now a trend towards accepting ISO 15189 as the standard for all accreditation bodies in order to provide uniformity of standards and cross-recognition. Examples of accreditation authorities include the CPA (College of American Pathologists) in the U.S., CPA (Clinical Pathology Accreditation) in the UK, NATA in Australia and HOKLAS for Hong Kong.
UK System
In the UK, pathology department accreditation is carried out by an independent non-profit making company known as CPA (Clinical Pathology Accreditation) which is jointly owned by several bodies including the Royal College of Pathologists. There are similar bodies for laboratory accreditation in other countries such as the that operated by the Colloege of American Pathologists in the US and NATA in Australia. In the past, the standards of these accreditation bodies varied.
Accreditation is an external audit of an applicant department’s organization and quality assurance program. The CPA defines standards for organization and performance of clinical pathology. Applicant departments assess themselves against those standards and fill in a form to indicate compliance with or, exemption from them and send this, together with details of their facilities and repertoire to CPA. At this stage, provisional accreditation is awarded if all seems to be in order. Some time later, the department is subjected to an on-site inspection. If all seems to be well, full accreditation is granted at this stage. Should the inspectors identify problems, full accreditation is withheld until the problems have been solved to the satisfaction of the CPA. If that is not achieved within a reasonable time, provisional accreditation lapses. The refusal of accreditation may be appealed by the applicant department, the CPA board’s decision will be binding. Fully and provisionally accredited departments will be put on the CPA register. Those with full accreditation will be reinspected at regular intervals, probably every 4 years. CPA inspectors will usually visit each department in pairs, each team consisting of a consultant or clinical scientist and a senior MLSO. This means that for an "average" NHS hospital with four pathology departments, eight inspectors will visit for one full day. In addition to on-site inspection of the laboratory, service users and hospital managers will be interviewed.
Standards of the UK CPA
There are 44 standards grouped under 6 headings, most of which are applicable to all pathology departments.
A. Organization and Administration
There is a document to describe the organization and appropriate overall scope of the laboratory service and a documented line of managerial accountability. There are formal meetings between senior laboratory staff and management to review the service, set objectives and make financial arrangements
B. Staffing and Direction
Each discipline is directed by a consultant pathologist or clinical scientist there are appropriate numbers of staff with the required training. There is a documented line of accountability for all staff to the head of department - all unqualified staff must be supervised. Job descriptions must be available for all staff and all staff must have a contract of employment Regular staff meetings should be held and all new staff are given a comprehensive induction program
C. Facilities and Equipment
There are a total of 10 standards under this heading. This include appropriate laboratory and office space, staff facilities, space for specimen reception and handling and specimen storage. There should be adequate lighting, heating, ventilation, gas and water drainage. A safe working environment must be available for which he safety officer is responsible. There are appropriate data storage, retrieval and communication facilities. There is appropriate properly maintained scientific equipment that should be properly maintained.
D. Policies and Protocols
There should be a user manual to help clinicians to provide the right sample, to use the service to the best effect and what help and advice is available. Laboratory forms should include unique patient identity and supporting information. Reports should be accurate, comprehensive and clinically relevant and should include adequate patient identity, time and date of collection, testing and reporting. A written record of all reagents, calibration and quality control material. There must be written protocols for the performance of each test including the appropriate preparation of equipment, sample reagents, calculation of results and interpretation of internal quality control performance. There is a written protocol for the normal reporting and oral reporting of results. There should also be protocols for the regular maintenance as well as the decontamination of all items of equipment and working space. A policy describing any out of hours service must be instituted. In hospitals, a nominated consultant microbiologists is responsible for institutional infection control.
E. Staff Development and Education
There is a written program of training for all members of staff, appropriately sited facilities, resources and facilities to attend external seminars, meetings and conferences, and a continuing education program. In addition there should be an established staff appraisal scheme.
F. Evaluation
The department must participate in all appropriate external quality assessment programs which should be widely publicized in the department. Clinical audit of the service should be continually carried and senior staff should regularly participate in the audit activities of other clinical specialties.
Audit in the Clinical Laboratory
The main thrust of the NHS reforms is to provide the best possible cost-effective service to the patient. The main effect is to run each part in the NHS like a commercial organization. Audit is an essential means of achieving such an aim and this is recognized by the financial resources made available by the Department of Health for it. A number of factors will ensure that audit will remain an essential part of the service;-
The audit commission is now responsible for the external audit of NHS activities
Purchaser pressure - purchasers (health authorities, local authorities, fund-holding GPs etc.) will be looking for evidence of high quality cost-effective services
Professional clinical audit by al health care professionals is consistent with the commitment to improve the quality of services to the patient.
Managerial - many aspects of medical audit are of interest to health managers who may use the results for the determination of managerial policies
Commercial - as laboratories increase their contact with commercial organizations e.g. by income-generating work, the customers may want to look for information about the quality of the service they intend to purchase.
Audit is an essential part of the quality assurance program of a laboratory. A quality assurance program covers all aspects of the service provided. It may include policies on the induction and training of new staff, staff development, laboratory manuals, safety policies, equipment maintenance etc. Audit is a means of assessing whether one is achieving one's stated objectives. There are five key questions in the audit process:
Financial audit and the work of the audit commission are undertaken by auditors from outside the laboratory. The medical virologist may have an important role in medical and laboratory audit.
Medical Audit
All doctors are required to participate in medical audit which is defined as the systematic critical analysis of the quality of medical care. The medical virologists may be involved as part of the multidisciplinary team which may cover topics such as infection control, appropriate use of the virology laboratory, antiviral usage.
Laboratory Audit
Laboratory audit is concerned primarily with the everyday aspects of the work of the department and is a means of providing feedback to both the users of the laboratory and its staff. Laboratory audit is usually organized internally although the NEQAS and Clinical Pathology Accreditation schemes can complement the in-house program of audit. A diagnostic virology may wish to examine the following areas by audit.
Request forms; are they easy to use? Are all relevant details provided by the user. For example, date of contact or onset would be particularly useful in the case of requests for rubella serology
Specimens; is the right specimen received at the right time? Are the appropriate investigations selected by the laboratory staff? It is essential that specimens for virus isolation should arrive at the laboratory as soon as possible after collection or else the chance of isolation will decrease. This is not so important for serological tests. The laboratory staff would have much more leeway in selecting tests in the general serology department than other departments.
Turn-around times for each request. Attempts should be made to monitor the turn-around time in each department and see whether improvements can be made. Virology laboratories generally have the worse reputation for turn-around times compared to other pathology laboratories.
Is the range of investigations available appropriate? The number of requests for a specific test and the positivity rate should be audited. Those tests for which requests which are rare and/or have a low positivity rate should be withdrawn.
Are the test methods being carried out according to standard operating procedures?
Safety policies and procedures. Every laboratory should have a comprehensive safety policy. Every single accident in the laboratory should be recorded and improvements made if necessary. The use of dangerous substances should be audited.
Efficient use of staff. Do senior staff perform duties that should or could be delegated to others. Efficient use of staff would be a much more important consideration in a small laboratory than a larger one. The training of all staff may be audited.
Purchasing of equipment, reagents, stationary and other items
Laboratory reports: are they precise and clear?
Storage of reagents and specimens
Complaints and corrective action taken
The audit process begins with the auditor drawing up an audit checklist compiled from the quality system manual of the part being audited. The auditor then checks compliance, non-compliance or possible non-compliance against this checklist and write a report. Corrective action requests may then be submitted as part of the report. The quality system itself can be audited by a non-technical person, whereas technical activities must be audited by a person with sufficient technical background. In general, it is better to have a series of small audits rather than a single large audit. Any faults identified by an audit should lead to immediate corrective action and appropriate changes in documentation, which should be discussed in management reviews.
![]()
![]()