10. Molecular Techniques
Molecular biology techniques for the direct detection of viral genomes in the specimen will play an increasingly important role in the clinical virology laboratory in the 21st century. Molecular techniques can be divided into two categories: those that do not involve amplification i.e. hybridization with nucleic acid probes, and those that involve amplification e.g. PCR, LCR, NASBA etc.
Nucleic Acid Probes
Nucleic acid probes are segments of DNA or RNA that have been labeled with enzymes, antigenic substrates, chemiluminescent moeities, or radioisotopes. They can bind with high specificity to complementary sequences of nucleic acid. Probes can be directed to either DNA or RNA targets and can be from 20 to thousands of bases long. The presence and the quantity of hybrids after hybridization is determined by the detection of the label. Probes are usually synthesized by one of the following three methods.
1. Oligonucleotide probes - these are usually less than 50 bases long and are synthesized chemically
2. PCR - will produce probes from 50 to several hundred bases long
3. Cloning - will produce probes several thousand bases long e.g. probe for complete HBV genome
Altering the stringency of the reaction will alter the sensitivity and specificity of the hybridization. Stringency relates to the number of mismatched base pairs that can be tolerated when two nucleic acid molecules come together to form a double stranded molecule. Stringency is affected by several variables, including the temperature, salt concentration, and the pH of the hybridization reaction. High stringency is achieved by using buffers of low salt concentration or by conducting the hybridization reaction and stringency washes at higher temperatures. The higher the stringency of reaction, the less likely it is for mismatched base pairs to stay together.
The hybridization reaction may be carried out completely in solution phase whereby both the target nucleic acid and the probe are free to interact in the reaction mixture. Solution hybridization has the advantages of being rapid to the carry and carry a higher sensitivity than solid phase hybridization. This is the approach taken by Abbott with their quantitative HVB-DNA assay. Nucleic acid bound to a solid surface are still available to participate in hybridization reactions. However, the sensitivity tends to be lower than that of liquid hybridization. However, this technique greatly facilitates the handling of multiple samples. The dot-blot and sandwich hybridization assays are commonly used in this respect. In situ hybridization assays, in which whole cells or tissue sections are put through the hybridization process has become an important research tool.
Despite having been around for many years, hybridization
assays are still not in common use in the clinical virology
laboratory. The main reason is that its sensitivity is not
usually higher than far simpler conventional virological
techniques such as cell culture and viral antigen detection.
Polymerase Chain Reaction
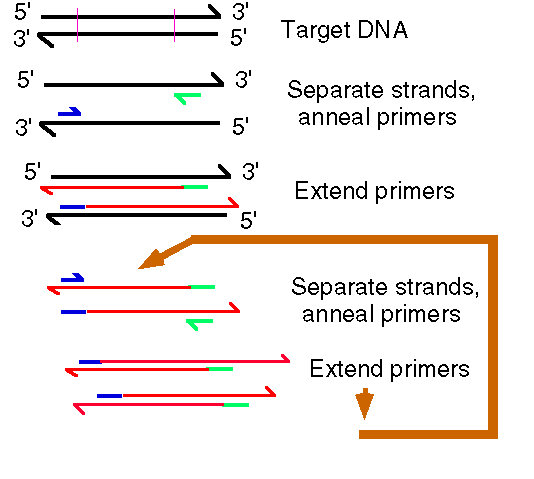
PCR allows the in vitro amplification of specific target DNA sequences by a factor of 106 and is thus an extremely sensitive technique. It is based on an enzymatic reaction involving the use of synthetic oligonucleotides flanking the target nucleic sequence of interest. These oligonucleotides act as primers for the thermostable Taq polymerase. Repeated cycles (usually 25 to 40) of denaturation of the template DNA (at 94oC), annealing of primers to their complementary sequences (50oC), and primer extension (70oC) result in the exponential production of the specific target fragment. Further sensitivity and specificity may be obtained by the nested PCR technique, whereby the DNA is amplified in two steps. In the first step, an initial pair of primers is used to generate a long sequence that contain the target DNA sequence. A small amount of this product is used in a second round of amplification, which employs primers to the final target DNA.
Schematic of Polymerase Chain Reaction
Detection of DNA sequence product of the PCR assay may be performed in several ways. The least sensitive and specific method is to size fractionate the reaction product on an agarose or acrylamide gel and stain the DNA with ethidium bromide. A more sensitive technique involves the attachment of DNA to a membrane through dot or slot-blot techniques followed by hybridization with a labelled homologous oligonucleotide probe. Alternatively, the PCR product may be probed directly by liquid oligomeric hybridization. However, these techniques provides no information on size of the amplified product and thus could not exclude the possibility that the product originated from a region of the human genome which exhibits homology with the target CMV sequence. The most sensitive and specific detection methods result from combining the size information of gel electrophoresis with the improved sensitivity and specificity of hybridization techniques. This may be achieved by gel electrophoresis followed by Southern transfer and hybridization, or through liquid oligomeric hybridization followed by gel electrophoresis.
Advantages of PCR:
Extremely high sensitivity, may detect down to one viral genome per sample volume
Easy to set up
Fast turnaround time
Disadvantages of PCR
Extremely liable to contamination
High degree of operator skill required
Not easy to quantitate results
A positive result may be difficult to interpret, especially with latent viruses such as CMV, where any seropositive person will have virus present in their blood irrespective whether they have disease or not.
The first three problems are being addressed by the arrival of commercial closed systems such as the Roche Cobas Amplicor which requires minimum handling. The use of synthetic internal competitive targets in these commercial assays has facilitated the quantification of results. Otherwise, the same problems remain with in-house PCR assays. The problem with contamination is particularly acute with nested PCR assays and they should be avoided in the clinical setting as far as possible. The sensitivity would normally be sufficient if one use a single PCR reaction followed by hybridization with a specific oligonucleotide probe. This is the approach taken by commercial assays. The fourth problem is more difficult to resolve but it is generally found that patients with active CMV disease has a much higher viral load than those who do not. Therefore, it is simply a case of finding the appropriate cut-off.
Other Amplification Techniques
Following the heels of PCR, a number of alternative in-vitro amplification techniques have been developed, of which some are now available commercially. Examples of these alternative techniques include ligase chain reaction (LCR), nucleic acid sequence based amplification/isothermal amplification (NASBA), strand displacement amplification, Qb Replicase method, and branched DNA probes. Of these techniques, LCR, NASBA and branched DNA are now available commercially in an automated or semi-automated format. A NASBA assay is available for the quantification of HIV-RNA (Organon), and an LCR assay is available for the detection of chlamydia (Abbott). Branched DNA assays are available for the detection of quantification of HIV-RNA, HBV-DNA, and HCV-RNA (Chiron).
With the exception of the branched DNA probe, all these techniques involve exponential amplification of either the target nuclei acid or the probe. Therefore, they are all as susceptible to contamination as PCR. The branched DNA system is really an intermediate between classical hybridization techniques and the newer in-vitro amplification techniques. It is not as sensitive as those techniques which involve exponential amplification but is considerably more sensitive than the classical hybridization techniques. Below is a brief summary of the features of the different amplification methods available.
![]()
![]()
![]()