Acyclovir
ANTIVIRAL_CHEMOTHERAPY
Vaccines have, to date, occupied the central position in attempts to control virus infections. Vaccines are relatively cheap and safe and the immunity is often lifelong. However, some viruses, for some reasons, are not fully amenable to this approach, such as influenza, retroviruses, herpesviruses, the slow viruses, rhinoviruses and arboviruses. Obstacles to the use of vaccines include (1) multiplicity of serotypes e.g.. rhinoviruses, togaviruses (2) antigenic change e.g.. influenza, retroviruses and (3) Latent infections. Only relatively recently have notable successes on a large scale been achieved with antiviral drugs such as acyclovir and AZT, in situations where no vaccine is available. However. acyclovir and AZT do not approach penicillin in their spectra of activity or degree of inhibition. They are more analogous to some of the first antibacterial agents such as salvarsan. No antiviral compound tested has been able to inhibit completely the replication of any virus and a proportion of viral particles always seems to be able to circumvent the drug-induced blockade.
A. The Chemistry of Antiviral Compounds
There are few restrictions on the types of molecules that inhibit virus replication, at least in the laboratory. They vary greatly in complexity and include natural products of plants, synthetic oligonucleotides, oligosaccharides, simple inorganic and organic compounds and nucleoside analogues. Examples of antiviral compounds in current use include:
Nucleoside analogues - thousands of analogues of naturally occurring nucleosides have now been synthesized and tested in the laboratory, initially as herpesvirus inhibitors and many now are retested as anti-HIV agents. In addition to purine and pyrimidine nucleosides, ara-, amino, aza-nucleosides or nucleotides have been synthesized. Even single atomic substitution may change an active to an inactive molecule.
Pyrophosphate analogues - forscarnet is an example of a pyrophosphate analogue. This specifically inhibits herpesvirus DNA polymerase at the pyrophosphate binding sites and it also has anti-HIV activity.
Amantidine molecules - amantidine is licensed for the treatment of influenza A infection. Addition of a methyl grouping (rimantidine) alters the pharmacological distribution of the drug and prevents entry to the brain, thus reducing the side effect described as "jitteriness"
The search for new antiviral compounds
All the antiviral drugs now known were discovered by random search in the laboratory. With developments in molecular biology and the advent of HIV, more attention is now being paid to the development of drugs designed to act on specific targets on the virus itself or in its replication. Attempts are being made to exploit data on viral nucleic acid sequences, X-ray crystallographic studies of viral proteins and enzymes. A recent example of the success of this new rational approach has been the discovery of potent neuraminidase inhibitors of influenza viruses.
Resistance of viruses to inhibitors
A disappointing feature of antiviral chemotherapy has been the failure so far of any antiviral molecule to inhibit virus replication completely. Antiviral activity tends to produce a 100 to 1000 fold reduction in virus titre which, although significant, still allows some infective particles to survive. This may have important consequences in immunocompromized patients who may be unable to eradicate any residual virus. It is not known for certain whether these virions are drug-resistant mutants or with different biologically or genetically from the major portions of the virus population.
B. Points of action of antiviral molecules
Thousands of compounds can inhibit viral replication in cell culture. In general, the more complex the regulatory mechanisms of a virus, the easier it is to find molecules that can inhibit it. It is often very hard to decide which of the compounds should be investigated further. A broad estimate of the ratios of the activity of antiviral compounds in cell culture, animal models and humans is 1000:10:1.
1. Cell-free virus
Few antiviral compounds inhibit or inactivate extracellular virus in vivo. An exception is the series of WIN compounds which bind to the external proteins of picornaviruses. These compounds bind to and fit within the canyons which exists on the surface of picornavirus virions and thereby stabilizing the particle and prevent uncoating.
2. Virus Adsorption
There is considerable theoretical interest in compounds able to block the adsorption of virus to susceptible cells. In the case of HIV, which binds specifically to CD4 receptors, short peptides have been synthesized to correspond with the sequence of the receptor binding site of CD4 molecule and with the binding protein gp120. These peptides should block the interaction of the receptor region and gp120 without interrupting other receptor functions of the CD4. The ant-HIV drug Maraviroc is an entry inhibitor. The chemokine receptor CCR5 is an essential co-receptor for most HIV strains. Maraviroc binds to CCR5, thereby blocking the HIV protein gp120 from associating with the cellular receptor and does making it unable to enter human macrophages and T cells. Because HIV can also use other coreceptors, such as CXCR4, an HIV tropism test such as a trofile assay must be performed to determine if the drug will be effective.
3. Virus entry and uncoating
Viruses such as influenza and certain flaviviruses enter by viropexis or engulfment. Immediately afterwards, while in a cytoplasmic endosome (vacuole), the virus catalyses fusion between the viral lipid-containing membrane and the membrane of the intracellular vacuole. The fusion is mediated by a sequence of hydrophobic amino acids or one of the glycoproteins of the virus. A compound that interrupts fusion would block virus replication at this early stage. In the case of influenza A, the fusion sequence on the HA molecule can only act after a structural 3- dimensional rearrangement of the HA molecule. This major change, whereby the HA trimer opens out like the petals of a flower, probably occurs only at a low pH5.5 found in lysosomal vacuoles. Amantidine appear to inhibit influenza A replication in part by raising the pH of the cytoplasmic vacuole, thus preventing virus-induced fusion and hence virus uncoating. Other enveloped viruses such as paramyxoviruses and HIV, enter cells by virus-induced fusion with the plasma membrane of the cell. This "fusion from without" may be susceptible to short peptides which may act on the fusion sequence extracellularly.
4. Transcription and translation of viral nucleic acids and release of virus
Most of the antiviral drugs now known act by inhibiting the replication or transcription of viral nucleic acids.
a. Inhibitors of herpes DNA polymerase - by far
the most amenable target for antiviral drugs is the
herpes simplex DNA polymerase. The most successful
antiviral compound yet developed is acyclovir inhibits
the function of this enzyme. The ideal antiviral drug
should (1) be taken up only into infected cells (2) the
actual inhibitory molecule should be generated inside the
infected cell by enzymatic activity (3) the inhibitor
should have a selective effect on a virus enzyme.
Acyclovir demonstrates all of the above characteristics.
b. Inhibitors of viral reverse transcriptase - AZT and the majority of other compounds act as chain terminators. AZT triphosphate binds to and inhibit virus RT more effectively than normal cellular DNA polymerases and so some antiviral specificity is achieved. However, the compound is certainly not comparable to acyclovir in terms of antiviral specificity. This is reflected in the toxicity of AZT in clinical practice. This cellular toxicity may be partly explained by the fact that normal cellular enzymes phosphorylate AZT and is thus activated in both infected and uninfected cells.
5. Translation
It may be possible to interfere with the viral mRNA itself. Small anti-sense oligonucleotides can be constructed which are complementary to specific genes, such as the rev gene. Fomivirsen (Vitravene) is a 21-base anti-sense oligonucleotide complementary to the early region 2 mRNA of CMV. It is approved for the local treatment of CMV retinitis in AIDS patients.
6. Assembly
HIV protease is required for the cleavage of the gag-pol
fusion protein. Inhibitors of this enzyme may therefore block the
assembly of HIV.
C. Some Commonly Used Antiviral Agents
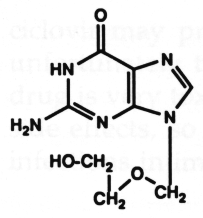
1. Acyclovir
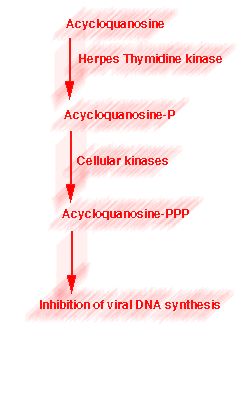
Acyclovir is a synthetic guanine nucleoside analogue. The initial step of phoshorylation to ACV monophosphate is preferentially carried out by viral thymidine kinase rather than cellular kinases. The monophosphate cannot leave infected cells so that more non-phosphorylated compound enters to make up for the depleted intracellular concentration, only to be converted to the monophosphate. In this manner, the drug accumulates in the herpes-infected cells rather than in the uninfected counterparts. The monophosphate is then phosphorylated to the di and tri-phosphate forms by cellular enzymes. ACV triphosphate is the pharmacologically active form of the drug. It inhibits herpes DNA polymerase with little effect on the host cell DNA polymerase. It also has some chain termination activity and thereby it behaves as a "suicide inhibitor"
Acyclovir |
|
Acyclovir resistant strains of HSV have mutations in either the viral thymidine kinase gene or the viral DNA polymerase. Acyclovir also has antiviral activity against other herpesviruses such as VZV, CMV and EBV, although the mechanism is not so well understood in these cases. Forscarnet is the preferred drug in the treatment of acyclovir-resistant strains.
2. Valacyclovir
Valacylovir is an ester of acyclovir that is well-absorbed. Its bioavailibility is 2-5* greater than acyclovir. It is used for the treatment and suppression of genital herpes infection.
3. Famciclovir
Famiciclovir is the prodrug of penciclovir which is the active form and a guanosine analog. It has a very high bioavailabiity of 77%. It is converted into penciclovir by a two step process. The first step occurs in the gut and the second step in the liver. It has a long half life in the gut. It has a higher affinity for HSV thymidine kinase than acyclovir but a lower affinity for HSV DNA polymerase than acyclovir. It acts as an inhibitor of viral DNA polymerase and also as a chain terminator. At present famciclovir is licensed for the treatment of shingles and the dosage is 250mg tds. It is also used for the treatment and suppression of genital herpes infection.
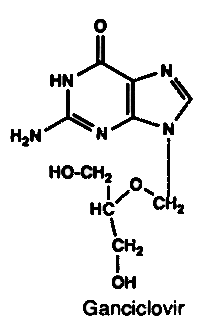
4. Ganciclovir
Ganciclovir is a guanine nucleoside chemically related to acyclovir. It acts as a chain terminator and subsequent termination of viral DNA replication. The active form is thought to be the triphosphate. CMV does not specify TK and the initial phosphorylation of ganciclovir is thought to be mediated by other cellular enzymes. Ganciclovir has potent in vitro activity against all herpesviruses, including CMV. It has some activity against other DNA viruses such as vaccinia and adenovirus. Ganciclovir is more active against CMV than acyclovir. Ganciclovir has been shown to be of value in treating severe CMV infections in the immunocompromized, especially in conjunction with hyperimmune immunoglobulin. Reversible neutropenia is the most frequent adverse reaction. Ganciclovir resistance has been reported in immunocompromised patients being treated for CMV disease and is thought to be due to lack of phosphorylation of the drug by CMV infected cells. A recent prospective study estimated that 8% of patients receiving ganciclovir for more than 3 months developed resistant CMV.
5. Ribavirin
Ribavirin is a synthetic triazole nucleoside and the active form is ribavirin triphosphate. It is not incorporated into the primary structure of DNA or RNA during cellular synthesis of nucleic acids. In the case of influenza viruses, it inhibits the 5' capping of viral mRNAs. It has also been shown to inhibit influenza viral RNA polymerase complex. It has further been postulated that ribavirin triphosphate inhibits several steps in viral replication and this phenomenon may explain the failure to detect viral isolates that are resistant to ribavirin. Ribavirin has been shown to possess activity against both DNA and RNA viruses in infected cells. It has been found to have activity against adenoviruses, herpesviruses, CMV. vaccinia. influenza A and B, parainfluenza 1, 2, 3, measles, mumps, RSV, rhinovirus. Ribavirin has made a major contribution to the therapy of children infected with RSV where is given as an aerosol in hospital. It has also been shown to be effective against influenza A and B. It has also been reported to be of value in the treatment of Lassa fever, hantavirus disease and hepatitis C.
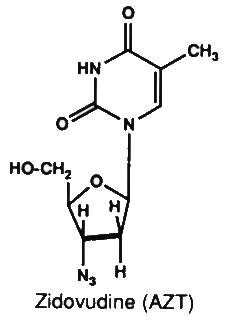
6. Zidovudine (AZT)
AZT is a synthetic analogue of thymidine. It requires
conversion to the triphosphate form by cellular enzymes. It
inhibits viral reverse transcriptase by acting as a chain
terminator. Viral reverse transcriptase is 100 times more
susceptible to inhibition by zidovudine triphosphate than host
cellular DNA polymerase. Once incorporated into the viral DNA
chain, viral DNA synthesis is terminated as no more
phosphodiester bonds could be formed. AZT is active in vitro
against many human retroviruses, including HTLV-I and HIV. AZT is
currently indicated for the management of patients with HIV
infection who have impaired immunity. (T4 cell count of 400- 500
or less) AZT has been clearly shown to prolong the life of
individuals infected with HIV. It has also been shown to be of
benefit for the treatment of symptomless individuals although
this is controversial.
7. Lamivudine
Lamivudine is a potent reverse transcriptase inhibitor. It is generally well tolerated by patients. It now usually forms an essential component in the combination therapy of HIV patients. Recently, it had been approved for the treatment of chronic hepatitis B.
8. Forscarnet
Forscarnet is a pyrophosphate analog and unlike nucleoside analogues, forscarnet does not need to be activated by cellular or viral kinases. Forscarnet binds directly to the pyrophoshate- binding sites of RNA or DNA polymerases. Forscarnet is difficult to use as it must be given continuously intravenously via an infusion pump. It is used for the treatment of CMV retinitis in AIDS patients receiving AZT therapy, as it does not have overlapping toxicity with AZT. It is also used in the treatment of AZT resistant HSV infections. Its major adverse effect is on renal function.
9. Amantidine
this compound inhibit the growth of influenza viruses in cell culture and in experimental animals. Amantidine is only effective against influenza A, and some naturally occurring strains of influenza A are resistant to it. The mechanism of action of amantadine is not known. It is thought to act at the level of virus uncoating. The compound has been shown to have both therapeutic and prophylactic effects. Amantidine significantly reduced the duration of fever (51 hours as opposed to 74 hours) and illness. The compound also conferred 70% protection against influenza A when given prophylactically. Amantidine can occasionally induce mild neurological symptoms such as insomnia, loss of concentration and mental disorientation. However, these symptoms quickly developed in susceptible individuals and cease when treatment is stopped. The therapeutic and prophylactic activity of amantidine is now generally accepted and numerous analogues of this compound have been prepared. Rimantadine is not as effective as amantadine but is less toxic. One factor that limits the usefulness of amantidine and rimantidine is the rapid development of resistance of these molecules in 30% of patients. These resistant mutants have been reported to be as capable of being transmitted and causing disease as the wild virus.
10. Zanamivir
The rational approach to drug design has led to the design of
several potent inhibitors of influenza neuraminidase of which two,
zanamivir and oseltamivir are licensed for the treatment of
influenza A and B infections. In clinical trials, both agents
have demonstrated efficacy with minimal side effects. Because of
its poor bioavailibility, Zanamivir must be given by inhalation
whilst oseltamivir can be given orally. Because selection of drug-resistant
mutants characterized by changes in NA requires prolonged passage
in tissue culture, development of zanamivir-resistant viruses is
not expected to occur readily in patients. The available
information suggests that mutants may be less stable in vivo. The
significance of changes in hemagglutinin remains to be evaluated.
Overall the NA family of anti-influenza drugs is showing
considerable promise; resistant variants do not occur readily and
may be biological cripples.
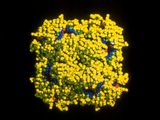 3-D structure of neuraminidase |
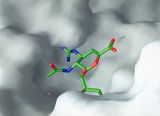 Neuraminadse inhibitor zanamivir fitting into the neuraminidase molecule |
11. Immunoglobulins
Immunoglobulins are available in three different formulations; intramuscular form, IVIG, and hyperimmune globulins against individual viruses. Immunoglobulins are more effective when used prophylactically rather than therapeutically. Currently, HNIG is used primarily for the prevention of hepatitis A. HNIG can also be given to non- immunized contacts of measles. Hyperimmune globulins are used for the postexposure prevention of hepatitis B, chickenpox and rabies. It has also been used in the treatment of arenavirus infections, Crimean-Congo haemorrhagic fever and Rift valley fever. CMV Ig is given prophylactically to seronegative recipients of kidneys from seropositive donors. The use of prophylactic CMV Ig in BMT patients is controversial. CMV IVIG is used in conjunction with ganciclovir in the treatment of CMV pneumonitis. IVIG is also used in the treatment of chronic enteroviral meningoencephalitis in children with agammablobinaemia.
The huge resources that had gone into HIV research had resulted in the development of a large number of anti-HIV agents. The speed of advance in this area is unprecedented in the history of medicine and is one of the greatest successes. To date with the correct therapy, there is no reason why an HIV-infected person cannot have the same life expectancy as a non-infected person. Therapy of HIV is complicated by the fact that the HIV genome is incorporated into the host cell genome and can remain there in a dormant state for prolonged periods until it is reactivated. However although it may or not be possible to actually eradicate the virus completely, it seems possible that the infection can be indefinitely contained so that an infected will die with HIV infection rather than from it.
Zidovudine (AZT) was the first anti-viral agent used for the treatment of HIV and was introduced in 1987. However, it became clear with Concorde study in 1994 that monotherapy with AZT did not provide durable efficiency and hardly made any dent in the mortality rate. In 1995, results of the European DELTA and the American ACTG 175 studies became available and showed that combination therapy with two nucleoside analogues were better than monotherapy with one alone. A further breakthrough occurred with the introduction of HIV protease inhibitors which were specifically designed against HIV protease and were shown to be the most potent anti-HIV effect to date. An early clinical trial reported that the use of oral ritonavir decreased HIV mortality from 38% to 22%. Combination therapy, otherwise known as HAART (highly active antiretroviral therapy) using two or three agents became available. The rationale for this approach is that by combining drugs that are synergistic, non-cross-resistant and no overlapping toxicity, it may be possible to reduce toxicity, improve efficacy and prevent resistance from arising. The final breakthrough occurred when David HO (Time Magazine Man of the Year 1996) finally elucidated the pathogenesis of HIV infection. He showed that far from being latent during the "latent phase" as previously thought, there is actually massive replication during this period. David Ho had coined the slogan "hit hard and early". The results of the new approach was seen quickly where within Within four years, between 1994 and 1998, the incidence of AIDS in Europe sank from 30.7 to 2.5/100 patient years i.e. to less than a tenth.
What is less hopeful is the possibility of ever eradicating HIV from the body i.e. complete cure. In the beginning, it is thought that continuous treatment for 3 years would be sufficient to eradicate all the remaining latently infected cells. However, the period of treatment required kept on being revised upwards as new data became available. The most recent estimate of eradication of all latently infected cells is 73.3 years. Therefore, it is clear it will not be possible to achieve a complete cure in the short term. Compliance is a major issue when therapy is expected to be life-long. There is clearly a great need to have formulations whereby the number of tablets to be taken per day is kept to a minimum. The development of side-effects with long term use is another issue.
As knowledge builds up on the risks and efficacy on various agents and regimens, recommendations on HIV are being continually revised. So now, instead of "hit hard and early", there is a shift towards "hit hard, but only when necessary". There is still a lot of debate on when to actually commence therapy. Two measures are used for determining whether to start HIV therapy: CD4 counts and viral loads. It is generally agreed that HIV therapy should be given when the CD4 count is below 200. Some experts would recommend treatment for any patient whose CD4 count is below 350. What is less clear are patients with CD4 counts of 300-500 and a modest viral loads. A decision to start therapy must be taken on an individual basis with the patient after thorough discussion and counseling.
1. Anti-Retroviral Agents
A. Nucleoside Reverse Transcriptase Inhibitor
B. Non-Nucleoside Reverse Transcriptase Inhibitor
C. HIV Protease Inhibitors
D. HIV Entry Inhibitors
Enfuvirtide (Fuzeon, ENF, T-20)
Maraviroc (Selzentry or Celsentri, MVC)
E. HIV integrase inhibitors
Raltegravir (Isentress, RAL)
Elvitegravir (EVG, part of the combination Stribild)
Dolutegravir (Tivicay, DTG)
There are a number of combination preparations on the market e.g. CBV (AZT+3TC), TZV (AZT+3TC+ABC), TVD (FTC+TDF), Kaletra (Lopinavir/ritonavir). The use of combination preparations will reduce the numbed of tablets that need to be taken each time.
2. Monitoring anti-HIV therapy
a. Viral Load
Initiation - viral load is now the preferred method of monitoring therapy. There should be >= 1 log reduction in viral load, preferably to less than 10,000 copies/ml HIV-RNA within 2-4 weeks after the commencement of treatment. If <0.5 log reduction in viral, or HIV-RNA stays above 100,000, then the treatment should be adjusted by either adding or switching drugs.
Monitoring - viral load measurement should be repeated every 4-6 months if patient is clinically stable. If viral load returns to 0.3-0.5 log of pre-treatment levels, then the therapy is no longer working and should be changed.
b. CD4 count
Initiation - within 2-4 weeks of starting treatment, CD4 count should be increased by at least 30 cells/mm3. If this is not achieved, then the therapy should be changed.
Monitoring - CD4 counts should be obtained every 3-6
months during periods of clinical stability, and more
frequently should symptomatic disease occurs. If CD4
count drops to baseline (or below 50% of increase from
pre-treatment), then the therapy should be changed.
c. Anti-HIV Drug Resistance Testing
Anti-retroviral drug resistance testing has become part and parcel of patient management in N. America and W. Europe. Many studies in treatment experienced patients have shown strong associations between the presence of drug resistance and failure of the antiretroviral treatment regimen to suppress HIV replication.
Genotypic Assays - genotypic assays detect drug
resistance mutations that are present in the relevant
viral genes (i.e. RT and protease). Some genotyping
assays involve sequencing of the entire RT and protease
genes, while others utilize oligonucleotide probes to
detect selected mutations that are known to confer drug
resistance. Genotyping assays can be performed relatively
rapidly, such that results can be reported within 1-2
weeks of sample collection. Interpretation of test
results requires an appreciation of the range of
mutations that are selected for by various antiretroviral
drugs, as well as the potential for cross-resistance to
other drugs conferred by some of these mutations.
Phenotypic Assays - phenotypic assays measure the
ability of viruses to grow in various concentrations of
antiretroviral drugs. Automated, recombinant phenotyping
assays have recently become commercially available with
turn-around times of 2-3 weeks; however, phenotyping
assays are generally more costly to perform compared with
genotypic assays. Recombinant phenotyping assays involve
insertion of the RT and protease gene sequences derived
from patient plasma HIV RNA into a laboratory clone of
HIV. Replication of the recombinant virus at various drug
concentrations is monitored by expression of a reporter
gene and is compared with replication of a reference
strain of HIV. The concentrations of drugs that inhibit
50% and 90% of viral replication (i.e. the IC50 and IC90)
are calculated, and the ratio of the IC50s of the test
and reference viruses is reported as the fold increase in
IC50, or fold resistance. Interpretation of phenotyping
assay results is complicated by the paucity of data on
the specific level of resistance (fold increase in IC50)
that is associated with failure of different drugs.
Use in clinical setting - resistance assays may be useful in the setting of virological failure on antiretroviral therapy. Recent prospective data supporting the use of resistance testing in clinical practice come from trials in which the utility of resistance tests were assessed in the setting of virological failure. The VIRADAPT and GART studies compared virological responses to antiretroviral treatment regimens when genotyping resistance tests were available to help guide therapy with those observed when changes in therapy were guided solely by clinical judgment. The results of both studies indicated that the short-term virological response to therapy was significantly greater when results of resistance testing were available. Similarly, a recent prospective, randomized, multicenter trial has shown that therapy selected on the basis of phenotypic resistance testing significantly improves the virological response to antiretroviral therapy, compared with therapy selected without the aid of phenotypic testing. Thus, resistance testing appears to be a useful tool in selecting active drugs when changing antiretroviral regimens in the setting of virological failure.
The rapidity of developments in anti-HIV therapy makes it virtually impossible for this site to keep up. For the latest information on HIV and anti-retroviral therapy, I recommend the HIV page at Medscape.com
There are 3 classes of interferons: alpha, beta and gamma. Interferon-a exists as at least 15 subtypes, the genes for which shows 85% homology. IFN b1 shows 30% homology with IFNa. IFNb2 is now known as IL-6 and shows no homology with alpha or b1 types. IFN gamma is also a lymphokine and shows no homology with the other types. IFNs mediate their actions through specific receptors at hormone like concentrations. Interferon inducible response elements in the cellular genome are activated. There are 2 main types of IFN receptors, one for alpha and beta1 and the other for gamma.
IFNs are released form many cell types in response to virus infection, dsRNA, endotoxin, mitogenic and antigenic stimuli. DsRNA appears to be a particularly important inducer. Usually, good IFN inducers are viruses that multiply slowly and do not block the synthesis of host protein early or markedly damage the cells. IFN is usually assayed by determining its effect on the multiplication of a test virus, usually vesicular stomatitis virus, a rhabdovirus. Viral strains capable of high IFN production give rise to autointerference in endpoint assays. In general IFN gamma differs from the others in that it is released as a lymphokine from activated T-cells and occasionally from macrophages.
1. Mechanism of Action
The antiviral effects of IFNs are exerted through several pathways;-
(1) Increased expression of Class I and Class II MHC glycoproteins, thereby facilitating the recognition of viral antigens by the immune system.
(2) Immunoregulatory effects - activation of cells with the ability to destroy virus-infected targets; these include NK cells and macrophages. IFNs appear to drive a shift from humoral to cellular immunity.
(3) Direct inhibition of viral replication: several mechanisms contribute to the third pathway.
production of specific inhibitory proteins eg. the Mx protein which has specific anti-influenza action. It is likely that more specific inhibitory proteins will be identified.
inhibition of viral processes such as penetration, uncoating and budding from infected cells have been reported.
in vitro studies with extracts of IFN-treated cells show that the main target of IFN action is translation, which is blocked by 2 mechanisms, both requiring the presence of minute amounts of dsRNA;-
(i). activation of a dsRNA dependent protein kinase - this phosphorylates and inactivates the translation initiation factor eIF-2. The phosphorylation freezes the initiation complex formed by eIF-2, GTP and met-tRNAf with the small ribosomal subunit and mRNA. Because eIF-2 cannot be recycled, protein synthesis is inhibited or stopped.
(ii). activation of 2-5 oligo A synthetases ® synthesis of 2-5A ® activates endonuclease L (itself induced by IFN) ® degradation of mRNA ® inhibition of protein synthesis.
The combination of cell growth inhibition and enhancement of CMI accounts for the antitumour effect of IFN.
2. Protective role in virus infections
A protective role of IFN in animals is suggested by many observations;-
In mice recovering from influenza virus infection, the titre of IFN is maximal at the time when the virus titre begins to decrease and before a rise in Abs can be detected. At this stage, the IFN titre in the animal is sufficient to protect them against the lethal action of a togavirus.
Administration of a potent antiserum to IFN markedly increases the lethality of mouse hepatitis virus infection.
Suckling mice, which are susceptible to coxsackievirus B1, produces little IFN in response to this virus, whereas adult mice, which are resistant, produces large amounts.
These studies suggest that IFN has a major protective role in at least some viral infections. Much depends on the dynamics of the disease.
3. Possible therapeutic use
Clinically, effective prophylaxis was demonstrated against rhinovirus infection of human volunteers, with decreased incidence of infection and reduction of symptoms. Contacts of infected patients can be protected by intranasal spray of large doses of IFN. Also reduced is CMV reactivation in seropositive patients undergoing kidney transplants. IFNs could theoretically be ideal antiviral agents, since they act on many different viruses and have high activity. However, their therapeutic value is limited by various factors: IFNs are effective only during relatively short periods and have no effect on viral synthesis that is already initiated in a cell. Moreover, at high doses they have serious toxic effects on the host.
Attempts to use exogenous IFN for the treatment of human viral
diseases had been met with limited success. IFN-a had prophylactic effect against influenza
infection during epidemics, and local administration lessens the
severity of respiratory diseases, IFN had also reported to be
successful in treating genital warts and juvenile laryngeal
papillomatosis. More recently, synthetic alpha-interferon was
licensed for use in the treatment of hepatitis B carriers and it
is also being used for the treatment of hepatitis C carriers with
chronic active hepatitis.
4. Interferon Therapy for Chronic HBV Carriers
It has been postulated that chronic carriage of HBV is due mainly to inadequate production of interferon and the failure of the body to respond to interferon in the presence of acute HBV infection. Two preparations of interferons are currently available: Alpha-Interfron (Intron A) and Peginterferon (Pegasys). In In early clinical trials, interferon therapy is associated with HBeAg loss in 30-40% of patients, and in approximately 10% lost HbsAg altogether. If a patient loses HBeAg loss during interferon therapy, HBsAg loss follows therapy in approximately 80% of patients followed for a decade. In addition, improved survival, complication-free survival, and a reduction in the frequency of hepatocellular carcinoma have been reported in those who responded to interferon. Interferon therapy is more effective in patients with low-level HBV DNA 100,000–40 million copies per mL, elevated ALT (esp if >200 IU/mL), immunocompetence, normal liver function (albumin, bilirubin and coagulation), and acquisition of infection in adulthood. Early studies suggested that the efficacy of interferon was low in patients with pre-core-mutant HBV infection (HBeAg negative strains), but recent observations have renewed interest in interferon for this indication. Emerging data on PEG interferon may result in the first line use of PEG products alone or in combination with oral agents. However, interferon requires inconvenient injection therapy, is associated with a lot of side effects, and is no better than lamivudine in terms eAg seroconversion. Morover, it isof limited value in certain subgroups although it is the only medication that offers a chance at a complete cure.
Interferon-alpha (Intron A) is given by injection several times a week for six months to a year, or sometimes longer. The drug can cause side effects such as flu-like symptoms, depression, and headaches. Approved in 1991 and available for both children and adults.
Pegylated Interferon (Pegasys) -
peginterferon is is modified form of interferon that has been approved for
the treatment of HBV and HCV. It has a similar but larger chemical
structure than interferon-alpha. This improves the efficiency of the drug so
that it only needs to be injected once weekly, usually for six months to a
year. The drug can cause side effects such as flu-like symptoms, depression
and other mental health problems. Approved May 2005 for adults.
5. Interferon therapy of Hepatitis C Carriers
Early studies indicate that interferon and ribavirin are effective of cases of acute and chronic hepatitis C. A combination of interferon and ribavirin may be useful. There is more experience in the use of interferon for the treatment of hepatitis C. The current recommendation is that interferon treatment may be considered in those with chronic active hepatitis who are at risk of progression to cirrhosis and HCC. The recommended regimen is 3 MU tds sc or im for 6 months. The response rate is around 50%. However, approximately 50% of responders relapse upon cessation of treatment. At present, it is not clear what factors predict response to interferon therapy. There is some data to suggest that older patients and those with established cirrhosis respond less well. There is growing evidence that the genotype of the infecting HCV determines the response to IFN. Most responders will have significant reduction of SGPT level within 2 months of interferon therapy. One may try a higher dose such as 5 or 10 MU in non-responders although it is not certain whether the higher doses work. At present, it is not clear what factors predict relapse after treatment. For those who relapse after treatment, they may be offered a second course and then put on maintenance therapy for 6 to 12 months. There is data to suggest that combination therapy with interferon and ribavirin is more effective than interferon alone. In fact, a pharmaceutical preparation of both these agents together is available for this purpose. It is now also routine to test for the HCV genotype before the commencement of Interferon/Ribavirin therapy. Genotypes 1 and 4 carry a poorer prognosis and response and typically these patients are treated for 48 weeks instead of 24 weeks for other genotypes.
![]()
![]()