![]()
Individual Methods
In this section, some commonly used virological methods will be discussed in further detail
Viruses are obligate intracellular parasites that require living cells in order to replicate. Cultured cells, eggs and laboratory animals may be used for virus isolation. Although embroyonated eggs and laboratory animals are very useful for the isolation of certain viruses, cell cultures are the sole system for virus isolation in most laboratories. The development of methods for cultivating animal cells has been essential to the progress of animal virology. To prepare cell cultures, tissue fragments are first dissociated, usually with the aid of trypsin or collagenase. The cell suspension is then placed in a flat-bottomed glass or plastic container (petri dish, a flask, a bottle, test tube) together with a suitable liquid medium. e.g. Eagle's, and an animal serum. After a variable lag, the cells will attach and spread on the bottom of the container and then start dividing, giving rise to a primary culture. Attachment to a solid support is essential for the growth of normal cells.
Primary and Secondary Cultures
Primary cultures are maintained by changing the fluid 2 or 3 times a week. When the cultures become too crowded, the cells are detached from the vessel wall by either trypsin or EDTA, and portions are used to initiate secondary cultures. In both primary and secondary cultures, the cells retain some of the characteristics of the tissue from which they are derived.
Cell Strains and Cell Lines
Cells from primary cultures can often be transferred serially a number of times. The cells may then continue to multiply at a constant rate over many successive transfers. Eventually, after a number of transfers, the cells undergo culture senescence and cannot be transferred any longer. For human diploid cell cultures, the growth rate declines after about 50 duplications. During the multiplication of the cell strain, some cells become altered in that they acquire a different morphology, grow faster, and become able to start a cell culture from a smaller number of cells. These cells are immortalized and have an unlimited life-span. However, they retain contact inhibition.
Cell Cultures
Cell cultures are separated into 3 types:-
Primary cells - prepared directly from animal or human tissues and can be subcultured only once or twice e.g. primary monkey or baboon kidney
Semi-continuous diploid cells - which are derived from human fetal tissue and can be subcultured 20 to 50 times e.g. human diploid fibroblasts such as MRC-5
Continuous cells - derived from tumours of human or animal tissue e.g. Vero, Hep2
Cell cultures vary greatly in their susceptibility to different viruses. It is of utmost importance that the most sensitive cell cultures are used for a particular suspected virus. Specimens for cell culture should be transported to the laboratory as soon as possible upon being taken. Swabs should be put in a vial containing virus transport medium. Bodily fluids and tissues should be placed in a sterile container.
Upon receipt, the specimen is inoculated into several different types of cell culture depending on the nature of the specimen and the clinical presentation. The maintenance media should be changed after 1 hour or if that is not practicable, the next morning. The inoculated tubes should be incubated at 35-37oC in a rotating drum. Rotation is optimal for the isolation of respiratory viruses and result in an earlier appearance of the CPE for many viruses. If stationary tubes are used, it is critical that the culture tubes be positioned so that the cell monolayer is bathed in nutrient medium.
The inoculated tubes should be read at least every other day for the presence of cytopathic effect. Certain specimens, such as urine and faeces, may be toxic to cell cultures that may produce a CPE-like effect. If toxic effects are extensive, it may be necessary to passage the inoculated cells. Cell cultures that are contaminated by bacteria should either be put up again or passed through a bacterial filter. Cell cultures should be kept for at least one to two weeks (longer in the case of CMV). Cell cultures should be refed with fresh maintenance medium at regular intervals or if required should the culture medium become too acidic or alkaline. When CPE is seen, it may be advisable to passage infected culture fluid into a fresh culture of the same cell type. For cell-associated viruses such as CMV and VZV, it is necessary to trypsinize and passage intact infected cells. Other viruses such as adenovirus can be subcultured after freezing and thawing infected cells.
|
|
|
|
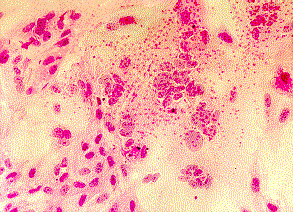
Cytopathic effects of enterovirus 71, HSV, and CMV in cell culture: note the ballooning of cells. (Linda Stannard, University of Cape Town, Virology Laboratory, Yale-New Haven Hospital)
|
|
|
Cytopathic effects of mumps and measles viruses in cell culture: note the formation of syncytia. (Courtesy of Linda Stannard, University of Cape Town, S.A.)
Influenza and parainfluenza viruses do not ordinarily induce CPE, however they possess haemagglutinins and thus the ability to absorb guinea pig RBCs as they bud from the cell. This phenomenon is known as haemadsorption. Commonly employed cell cultures include primary monkey kidney, LLC-MK2 and MDCK cells. The cell cultures are incubated with a suspension of guinea pig RBCs at 4oC or RT for 30 minutes. The unabsorbed RBCs are then removed and the cell sheet observed microscopically for the presence of haemadsorption. Presumptive identification of virus isolates can usually be made on the basis of the type of CPE, haemadsorption, and selective cell culture susceptibility. For final identification, immunofluorescence, neutralization, haemadsorption inhibition, lectron microscopy, or molecular tests are normally carried out.
Advantages of cell culture for virus diagnosis include relative ease, broad spectrum and sensitivity. It is limited by the difficulty in maintaining cell cultures, variability of cell cultures. Contamination by endogenous viral agents such as SV40, mycoplasma and bacteria may occur. Another problem in isolating certain viruses, especially myxo and paramyxo viruses is the presence of inhibitory substances or antibodies in the calf serum used in the cell culture media. Using fetal calf serum reduces this problem but adds to the expense.
Rapid Culture Techniques e.g. DEAFF test
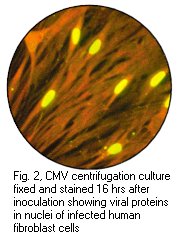
One of the most significant contributions to rapid diagnosis has been the application of centrifugation cultures to viral diagnosis. For a number of years, it has been recognized that low-speed centrifugation of specimens onto cell monolayers enhances the infectivity of certain viruses as well as chlamydia. The cell culture is stained by monoclonal antibodies for the presence of specific viral antigens 24-48 hours later. The best known example of this technique is the DEAFF test used for the early diagnosis of CMV infection. In the DEAFF test, the specimen is inoculated onto human embroyonic fibroblasts and then spun at a low speed. After a period of 24-48 hours, the cells are then stained by monoclonal antibodies against CMV early antigen. Therefore a rapid diagnosis of CMV infection can be made without having to wait 1-3 weeks for the CPE to appear.
|
|
|
Left: Haemadsorption of red blood cells onto the surface of a cell sheet infected by mumps virus. Also note the presence of syncytia which is indistinguishable from that of RSV (Courtesy of Linda Stannard, University of Cape Town). Right: Positive CMV DEAFF test. (Virology Laboratory, Yale-New Haven Hospital)
Susceptible Cell Lines
Herpes Simplex Vero Hep-2, human diploid (HEK and HEL),human amnion
VZV human diploid (HEL, HEK)
CMV human diploid fibroblasts
Adenovirus Hep2, HEK,
Poliovirus MK, BGM, LLC-MK2, human diploid, Vero, Hep-2,Rhadomyosarcoma
Coxsackie B MK, BGM, LLC-MK2, vero, hep-2
Echo MK, BGM, LLC-MK2, human diploid, Rd
Influenza A MK, LLC-MK2, MDCK
Influenza B MK, LLC-MK2, MDCK
Parainfluenza MK, LLC-MK2
Mumps MK, LLC-MK2, HEK, Vero
RSV Hep-2, Vero
Rhinovirus human diploid (HEK, HEL)
Measles MK, HEK
Rubella Vero, RK13
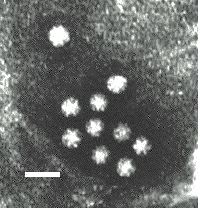
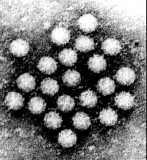
2. Electron Microscopy
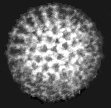
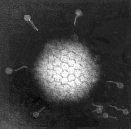
Virus diagnosis by electron microscopy relies on the detection and identification of viruses on the basis of their characteristic morphology. A major advantage of virus diagnosis by EM is the ability to visualize the virus. By identifying the virus directly, it is possible to perform an examination without a preconceived concept of the aetiological agent, in contrast with those assays which require a specific viral probe. Speed is another advantage of EM as the specimen can be processed within minutes of receipt and thus EM can be used as a rapid diagnostic method. On the other hand, the main disadvantage of EM is its inability to examine multiple specimens coincidentally. Secondly, there must be a minimum number of virus particles present (around 106 virus particles per ml for detection) Some viruses such as SRSV may give a non-distinct morphological appearance which may make detection very difficult. Finally, EM is a very expensive service to provide and requires highly skilled personnel. EM has found a particular niche in the detection of fastidious gastroenteritis viruses such as rota, adeno, astro, Norwalk, and Caliciviruses. It is also used for the rapid diagnosis of herpesvirus infection. It is occasionally used for the diagnosis of human papillomavirus infections and infections by members of the poxvirus family. In addition EM may be used to confirm the results of virus isolation by cell culture such as for parainfluenza viruses.
There are two types of EM methods;- direct or immunoelectron microscopy (IEM). With direct methods, negative staining is normally used which requires little special equipment, in contrast to thin sectioning techniques. The specimens may be used directly or the virus particles may be concentrated before negative staining. Several methods are available for concentration, including differential centrifugation, ammonium persulphate precipitation, and agar diffusion method. Fomvar coated copper grids are used. Immunoelectron microscopy is a means of increasing the sensitivity and specificity of EM and is particularly useful in the following situations;-
The number of virus particles present is small.
Many different viruses have different morphology e.g. herpesviruses and picornaviruses. IEM may identify the virus
In an outbreak situation where the pathogens responsible has been identified so that it may be useful to go back to look at the negative specimens again with IEM.
There are 2 types of IEM, simple IEM, where the specimen is
incubated with specific antibody before staining in the hope that
the antibody will agglutinate the specimen, and solid phase IEM
(SPIEM), where the copy grid is coated with specific antibody
which is used to capture virus particles from the specimen.
|
|
|
|
|
Electronmicrographs of viruses commonly found in stool specimens from patients suffering from gastroenteritis. From left to right: rotavirus, adenovirus, astroviruses, Norwalk-like viruses. (Courtesy of Linda M. Stannard, University of Cape Town, http://www.uct.ac.za/depts/mmi/stannard/emimages.html)
The complement fixation test (CFT) was extensively used in syphilis serology after being introduced by Wasserman in 1909. It took a number of decades before the CFT was adapted for routine use in virology. CFT meet the following criteria; it is convenient and rapid to perform, the demand on equipment and reagents is small, and a large variety of test antigens are readily available. However, there is now a trend to replace the CFT with more direct, sensitive and rapid techniques, such as RIAs and EIAs. Although CFT is considered to be a relatively simple test, it is a very exacting procedure because 5 variables are involved. In essence the test consists of two antigen-antibody reactions, one of which is the indicator system. The first reaction, between a known virus antigen and a specific antibody takes place in the presence of a predetermined amount of complement. The complement is removed or "fixed" by the antigen-antibody complex. The second antigen-antibody reaction consists of reacting sheep rbc with haemolysin. When this indicator system is added to the reactants, the sensitized rbcs will only lyse in the presence of free complement. The antigens used for CFT tend to be group antigens rather than type-specific antigens. In order for the CFT to be set up correctly, the optimal concentration of haemolytic serum, complement, and antigen should be determined by titration. The following is a protocol for setting a complement fixation test.
a. Titration of haemolytic serum and complement
Dilutions of complement with 20% difference in concentration are made from 1:30 to 1:279. The following dilutions of haemolytic serum are made: 1:400, 1:800, 1:1600, 1:2000, 1:2400, 1:2800, 1:3200.
The following controls are required:
cell control - unsensitized cells only
complement control - complement at different concentrations and unsensitized cells
haemolytic serum control - sensitized cells only at different concentrations of haemolytic serum
The optimal sensitizing concentration (OSC) of haemolytic serum is the dilution which gives the most lysis with the highest dilution of complement. One haemolytic dose of complement (HD50) is the dilution that gives 50% lysis at the OSC of haemolytic serum. 3 HD50 of complement is used for the CFT
b. Titration of antigen and antibody
Antigen at dilutions of 1:2 to 1:512 is titrated against positive serum control. The following controls are incorporated:
antigen control - antigen at different concentrations, complement and sensitized cells
antibody control - antiserum at different concentrations, complement and sensitized cells
cell control well - sensitized cells only
complement back titration
The optimal dose of the antigen is the highest dilution of antigen that gives 75% or more fixation with the highest dilution of antibody.
c. CFT proper
In the CF proper, the haemolytic serum is used at the optimal sensitizing concentration, the complement at 3HD50, and each individual antigen at the optimal dose. Patients' sera should be inactivated at 56oC for 30 minutes before. Screening is usually carried out at around 1:10 in VBS, if positive, the serum is retested with doubling dilutions of 1:10 to 1:80. Control plates should be prepared with all the antigens used included. The following controls should be present;-
Serum control - serum and complement only, to detect any anticomplementary activity in the serum
Antigen control - antigen and complement only, to detect any non-specific reaction between antigen and complement.
Complement back titration - to check that the complement is used at the correct strength
Cell control - sensitized cells only, to check that the cells were suitable for use.
All controls should show complete lysis and in the complement back-titration, the reading should be 0 at the second well and 1 to 2 at the third well. The highest dilution of patient serum that still shows a reading of 3 or 4 is the CF titre. Diagnosis of a recent infection is usually made by the detection of a fourfold or greater increase in titre or by the detection of a high antibody titre from a single specimen (1:80 or above). Where antigens used are grown from yolf sac, the sera should be tested against control yolk sac antigen in order to exclude the possibility of non-specific reaction.
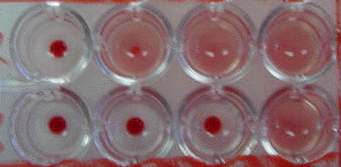
Complement Fixation Test in Microtiter Plate. Rows 1 and
2exhibit complement fixation obtained with acute and convalescent
phase serum specimens, respectively. (2-fold serum dilutions were
used) The observed 4-fold increase is significant and
indicates infection.
Advantages of CFT
Ability to screen against a large number of viral and bacterial infections at the same time.
Cheap
Disadvantages of CFT
Not sensitive - cannot be used for immunity screening
Time consuming and labor intensive
Often non-specific e.g. cross-reactivity between HSV and
VZV
4. Haemagglutination Inhibition Test
A wide variety of different viruses possess the ability to agglutinate the erythrocytes of mammalian or avian species. The actual animal species whose erythrocytes could be agglutinated depends on the actual virus. Examples of viruses which could haemagglutinate include influenza, parainfluenza, adenoviruses, rubella, alphaviruses, bunyaviruses, flaviviruses and some strains of picornaviruses. Antibodies against the viral protein responsible for haemagglutination can prevent haemagglutination; this is the basis behind the haemagglutination-inhibition test (HAI). The specificity of the HAI test varies with different viruses. With some viruses such as influenza A, the haemagglutination antigen is the same as the antigen responsible for virus adsorption and thus virus neutralization, and therefore the HAI test is highly specific for the different strains of the virus. With other viruses, the HAI test is less specific eg. flaviviruses, where HAI antibodies against one flavivirus may cross-react with other related flaviviruses. HAI tests are more sensitive than complement-fixation tests but are less sensitive than EIAs and RIAs.
The HAI test is simple to perform and requires inexpensive equipment and reagents. Serial dilutions of patient's sera are allowed to react with a fixed dose of viral haemagglutinin, followed by the addition of agglutinable erythrocytes. In the presence of antibody, the ability of the virus to agglutinate the erythrocytes is inhibited. The HAI test may be complicated by the presence of non-specific inhibitors of viral haemagglutination. and naturally occurring agglutinins of the erthrocytes. Therefore, the sera should be treated before use or false positive or negative results may arise. HAI tests are widely used for the diagnosis of rubella and influenza virus infections. The following is a brief description of the HAI test for rubella.
For rubella HAI testing, one day old chick or goose erythrocytes are used. Bovine albumin veronal buffer (BAVB) is used as the diluent. The HAI test should be carried out using 4 haemagglutination units of rubella antigen. The actual concentration of antigen required should be determined before each HAI test by carrying out a rubella antigen titration from 1:2 to 1:1024. One HA unit is defined as the highest dilution of antigen that gives complete haemagglutination of cells.
In the actual HAI test, the patients' sera are diluted in BAVB from 1:8 to 1:1024. Either V-shaped or U-shaped 96 -well microtitre plate may be used. Non-specific inhibitors of viral haemagglutination may be removed by the treatment of sera before testing by kaolin, RDE, potassium periodate (KIO) or by heat inactivation. Non-specific agglutinins for erythrocytes may be removed by the addition of erythrocytes to the sera prior to testing to allow the erythrocytes to absorb the non-specific agglutinins. This procedure may be carried out for each serum before testing or may be carried out for sera which had shown agglutination in the serum control wells (serum and erythrocytes only) in a previous HAI test. 4HA of rubella antigen is then added to each well containing diluted test sera except for the serum control wells. A back titration of rubella antigen should be incorporated into the test from 4 HA units to 0.25 HA units. The plate is then allowed to stand at room temperature for 60 minutes after which either 0.5% goose cells or 0.4% chick cells are added to each well and incubated at 4oC for 60 minutes. The plate is then read.
The erythrocytes only control should show a button at the bottom of the well. The serum controls for each serum should show the absence of agglutination. The haemagglutinin back titration should show agglutination at 4, 2 and 1 HA units. A fourfold or greater rise in HAI antibody between acute and convalescent phase sera is indicative of a recent rubella infection.
The advantages of HAI tests are that they are relatively easy and inexpensive to perform. The disadvantages are that HAI tests are not as sensitive as EIAs or RIAs, the actual reading of results is subjective and the reagents should be fresh or else abnormal agglutination patterns may arise which makes the reading and interpretation of the test very difficult. As a result the HAI test for rubella had been replaced by more sensitive and reliable EIA and RIA tests for rubella IgG in many virus diagnostic laboratory
ELISA was developed in 1970 and became rapidly accepted. A wide variety of assay principles can be used in ELISA techniques. Currently the most important ones are;-
Competitive methods
Sandwich methods
Antibody capture methods
Competitive methods
One component of the immune reaction is insolubilized and the other one labeled with an enzyme. The analyte can then be quantified by its ability to prevent the formation of the complex between the insolublized and the labelled reagent. Advantages of this approach are that only one incubation step is necessary and that the "prozone effect" at high analyte concentrations cannot occur. Disadvantages are that the concentration range in which the analyte can be quantified without sample dilution is rather narrow and that the antigen or antibody (in cases where either may be present in a sample e.g. hepatitis B) produce the same response, and can therefore cannot be distinguished in a one step assay.
Sandwich (Indirect) methods
The method in which the same component of the immune reaction (e.g. the antibody) is used in the insolubilized and the enzyme labelled form. The other component, the analyte (i.e. the antigen in the sample forms a bridge between the two reagents.)
The method in which one component (usually the antigen) is used in an insolubilized form to bind the analyte from the sample (the antibody),which is subsequently determined by addition of labelled second antibody against the same class of antibody as the analyte antibody or protein A.
In principle, quantification can be achieved over an extremely wide analyte concentration range in such sandwich methods. The "prozone effect" can be avoided in the following ways;- (i) using sequential incubation steps for sample and label, or (ii) by using monoclonal antibodies. Modification of the test in (2) so that antibodies of a specific class such as IgM, can give spurious results if antibodies from other immunoglobulin classes are also present in the sample. Also RF ( rheumatoid factor ) is known to be a potentially interfering factor.
Sandwich inhibition methods
The sample containing the analyte (usually antibody) is pre-incubated with a fixed amount of its binding partner (ie. the antigen ) after which the remaining amount of antigen is determined in a sandwich assay. These methods usually are complicated and have a limited measuring range. The method allows for simultaneous detection of antibody or antigen, if either of these 2 analytes is present in the sample.
Antibody capture methods
These methods used to detect antibodies of specific immunoglobulin subclasses, by first reacting the sample with e.g. insolubilized anti-IgM,and subsequently with either enzyme labelled antigen followed by enzyme linked antibody. Neither antibodies from other immunoglobulin subclasses nor rheumatoid factor interfere significantly in such assays. They are widely used for the diagnosis of acute infections by IgM detection. These assays may be used for detecting IgG and IgA. Considering trends towards simplification of assays and quantification of analytes over a wide concentration range. It must be expected that competitive and sandwich inhibition methods will decrease in importance, and the sandwich and antibody capture methods will be the main assay principles in the future.
Assay Characteristics
The use of monoclonal antibodies has lead to many improvements in ELISA systems.
Higher sensitivity ;- either by selection of antibodies with a extremely high affinity, or by reduction of the height and variability of the background reaction, which makes very low concentrations of analyte more readily detectable.
Higher specificity ;- by avoiding the presence of any antibody in the assay system with specific reactivity against non-analyte epitopes, and by selecting combinations of monoclonal antibodies which may further increase specificity.
Higher practicality ;- e.g. by introducing simultaneous incubation of label, solid phase and sample without risk of "prozone effect".
The enzyme label ;- Most of the assays employ horse-radish peroxidase, alkaline phosphatase, or B-D-galactosidase. The most interesting recent developments has been in new methods to detect these enzymes rather than the use of new enzymes. Fluorimeters were introduced in 1984 for the detection of alkaline phosphatase and B-D-galactosidase. Methods are available to detect horse radish peroxidase by means of chemilumininescence. Fluorimetric and luminometric methods offer higher sensitivity and a wider measuring range than conventional spectrometry. TMB is gradually replacing mutagenic substrates such as OPD, leading to increased sensitivity and safety.
Microplate ELISA: coloured wells indicate reactivity. The darker the colour, the higher the reactivity
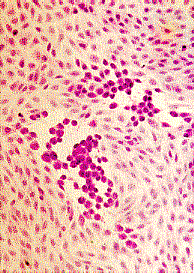
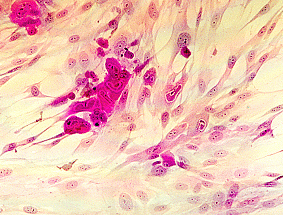
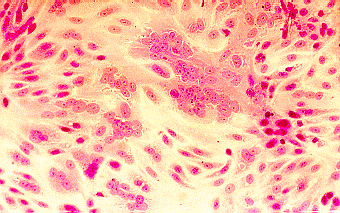
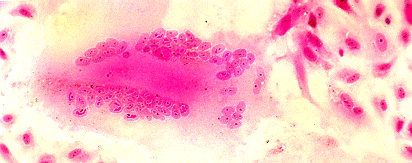
Single radial haemolysis (SRH) is routinely used for the detection of rubella-specific IgG. Many sera can be examined simultaneously for evidence of immunity by this technique. SRH has been shown to be both sensitive, specific, and reliable. SRH plates are usually prepared in the laboratory using commercially available reagents, since the short shelf life of the gels makes commercial distribution difficult. Test sera are placed in wells on a plate containing agar with rubella antigen-coated rbc and complement. The presence of rubella-specific IgG is detected by the lysis of rubella antigen-coated RBC. The zone of lysis around the well is dependent on the level of specific antibody present. Besides screening for immunity, SRH can also be used for the diagnosis of acute infection where an increase in the zone size of haemolysis can be demonstrated between acute and convalescent sera. SRH has also been developed for other virus infections such as mumps. However, its main use remains in the screening for immunity against rubella.
SRH plates are prepared as follows. 1% molten agarose is used. Rubella antigen-sensitized sheep rbcs is added to the test plate and unsensitized sheep rbcs to the control plate. Complement is then added to the plates and the molten agarose is allowed to set. The plates are then stored at 4oC until use. Wells are then cut on the test and control plates and serum is added to one well on the test plate and a corresponding well on the control plate. A negative control, a 15 IU/ml control, and a high positive control are used. The plates are then incubated in a moist chamber at 37oC overnight.
The size of the zone of haemolysis around a well containing test serum is compared to that of the 15 IU/ml control. If the zone size is greater than the 15 IU/ml control, then the person is considered to be immune. No zone of non-specific haemolysis should be present on the control plate. Hazy zones of haemolysis may also occur when acute phase sera are tested due to the presence of low avidity antibodies. Some women fail to produce antibody level of greater than 15 IU/ml even after several vaccinations, therefore many laboratories consider women with a well- documented history of more than one vaccination to be immune, if antibodies are detected by another assay.
Besides SRH, other tests such as ELISA and latex agglutination are also widely used for the screening of rubella antibodies. Sera from patients whose immune status is difficult to determine by SRH may give a clear-cut result with ELISA. Latex agglutination tests have the advantage of speed and simplicity and the technique is also very sensitive. Therefore LA is now used as the screening test for rubella antibodies in many laboratories in preference to SRH.
|
|
|
Single Radial Haemolysis for rubella antibody. Test-plate (L),
Control Plate (R). The well in the middle of the plate contains
the 15 miu/ml control serum. Note the clear zone of lysis
surrounding the well on the test plate which is absent on the
control plate. Specimens which gives a zone of lysis equal or
greater than the 15 miu/ml control well are regarded as positive
for rubella antibody. Should a zone of similar size is present on
the control plate, then the result is not valid.
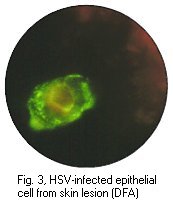
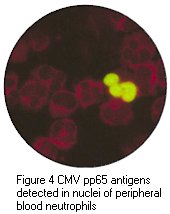
Immunofluorescence (IF) is widely used for the rapid diagnosis of virus infections by the detection of virus antigen in clinical specimens, as well as the detection of virus-specific IgG or IgA or IgM antibody. The technique makes use of a fluorescein- labelled antibody to stain specimens containing specific virus antigens, so that the stained cells fluoresces under UV illumination. In the case of direct IF, the specimen is probed directly with a specific labelled antibody against a particular virus antigen. In the case of indirect IF, the specimen is first probed with a non-labelled specific antibody, followed by a labelled antibody against the first antibody. Direct or indirect IF can be used for the detection of virus antigen, whereas indirect IF is virtually always used for the detection of antibody. Indirect IF possess the advantage of an extra amplification step for the signal, however, it requires an extra step in comparison to direct IF.
Detection of viral antigens
IF is most commonly used for the detection of respiratory viruses in respiratory specimens. Nasopharyngeal aspirates are the best specimens to use and is usually collected from babies less than 12 months old. However, there are no reasons why nasopharyngeal aspirates cannot be collected from older children and adults. A number of respiratory viruses can be detected by direct or indirect IF, including RSV, influenza A and B, adenoviruses and parainfluenza viruses. However, the sensitivities vary greatly between different viruses. The method is most useful in the case of RSV where antiviral treatment is available for severely ill babies. IF is also widely used for the detection of HSV infections, from vesicle lesions and brain lesions, for VZV and CMV infections. However in the case of CMV infection, the sensitivity of IF on clinical specimens directly is low, with the possible exception of the CMV antigenaemia test.
A typical indirect IF procedure for the detection of viral antigens is as follows;- cells from the clinical specimen are immobilized onto individual wells on a slide. Specific polyclonal or monoclonal sera is then added to each well and the slide is incubated at 37oC for 30 to 60 minutes. The slide is then washed 3 times for 5 minutes each with PBS and fluorescein labelled antibody against the first antibody is added. The slide is further incubated at 37oC for 30 to 60 minutes and washed again. The slide is then prepared for microscopy. Specific monoclonal or polyclonal sera raised against the viral antigen can be used. Monoclonal sera offer the advantage of increased sensitivity and specificity. However, one must be certain that it can detect all the different strains of the virus.
IF is highly dependent on the quality of the specimen. In many instances it has proved to be more sensitive than equivalent EIAs. This is because a firm diagnosis can be made on the identification of a few cells only that contain fluorescence of the right colour and with the correct antigen distribution. One of the criticisms of IF is that it is labor intensive and requires highly skilled staff for the reading the specimen.
|
|
|
|
Positive immunofluorescense tests of HSV antigen from epithelial cell and CMV pp65 antigen from peripheral blood neutrophils. (Virology Laboratory, Yale-New Haven Hospital). Right: Positive immunofluorescense test of rabies virus antigen (CDC)
Detection of viral antibodies
IF is probably the simplest serological assay to set up. It simply requires virally infected cells that express viral antigens and a fluorescein-labelled antiserum against human immunoglobulin. IF can be used to detect IgG, IgM and IgA. Being very easy to set up, it is often the first and only serological assay available for newly discovered viruses, in particular arboviruses. IF is extensively used for the diagnosis of EBV infections and is also routinely used for other viruses such as VZV.
8. Neutralization
Neutralization of a virus is defined as the loss of infectivity through reaction of the virus with specific antibody. Virus and serum are mixed under appropriate condition and then inoculated into cell culture, eggs or animals. The presence of unneutralized virus may be detected by reactions such as CPE, haemadsorption/haemagglutination, plaque formation, disease in animals. The loss of infectivity is bought about by interference by the bound Ab with any one o the steps leading to the release of the viral genome into the host cells. There are two types of neutralization;-
Reversible neutralization - The neutralization process can be reversed by diluting the Ab-Ag mixture within a short time of the formation of the Ag-Ab complexes (30 mins). It is thought that reversible neutralization is due to the interference with attachment of virions to the cellular receptors eg. the attachment of the HA protein of influenza viruses to sialic acid. The process requires the saturation of the surface of the virus with Abs.
Stable neutralization - with time, Ag-Ab complexes usually become more stable (several hours) and the process cannot be reversed by dilution. Neither the virions or the Abs are permanently changed in stable neutralization, for the unchanged components can be recovered. The neutralized virus can be reactivated by proteolytic cleavage. Stable neutralization has a different mechanism to that of reversible neutralization. It had been shown that neutralized virus can attach and that already attached virions can be neutralized. The number of Ab molecules required for stable neutralization is considerably smaller than that of reversible neutralization, Kinetic evidence shows that even a single Ab molecule can neutralize a virion. Such neutralization is generally produced by Ab molecules that establish contact with 2 antigenic sites on different monomers of a virion, greatly increasing the stability of the complexes. An example of stable neutralization is the neutralization of polioviruses, whereby, the attachment of the antibody to the viral capsid stabilizes the capsid and inhibits the uncoating and release of viral nucleic acid.
Viral evolution must tend to select for mutations that change the antigenic determinants involved in neutralization. In contrast, other antigenic sites would tend to remain unchanged because mutations affecting them would not be selected for and could even be detrimental. A virus would thus evolve from an original type to a variety of types, different in neutralization (and sometimes in HI) tests, but retaining some of the original mosaic of antigenic determinants recognizable by CFTs. Because of its high immunological specificity, the neutralization test is often the standard against which the specificity of the other serological techniques is evaluated.
Before the neutralization test is carried out, the known components that are to be used must be standardized. To identify a virus isolate, a known pretitred antiserum is used. Conversely, to measure the antibody response of an individual to a virus, a known pretitred virus is used. To titrate a known virus, serial tenfold dilutions of the isolate is prepared and inoculated into a susceptible host system such as cell culture or animal. The virus endpoint titre is the reciprocal of the highest dilution of virus that infects 50% of the host system eg. 50% of cell cultures develop CPE, or 50% of animals develop disease. This endpoint dilution contains one 50% tissue culture infecting dose (TCID50) or one 50% lethal dose (LD50) of virus per unit volume. The concentration of virus generally used in the neutralization test is 100 TCID50 or 100 LD50 per unit volume.
The antiserum is titrated in the neutralization test against its homologus virus. Serial twofold dilutions of serum is prepared and mixed with an equal volume containing 100TCID50 of virus. The virus and serum mixtures are incubated for 1 hour at 37oC. The time and temperature for incubation varies with different viruses. The mixtures are then inoculated into a susceptible host system. The endpoint titration contains one antibody unit and is the reciprocal of the highest dilution of the antiserum protecting against the virus. Generally 20 antibody units of antiserum is used in the neutralization tests.
Rubella reinfection can occur, especially in those whose immunity were induced by vaccination rather than by natural infection. However, reinfection by rubella during the first trimester of pregnancy is thought to pose minimal risks to the fetus. Cases of CRS arising from rubella reinfection have rarely been reported and termination of pregnancy is not recommended. Therefore, it is important to distinguish reinfection from primary infection by rubella during the first trimester of pregnancy. In the absence of reliable confirmatory tests, needless abortions may result.
Rubella specific IgM can be detected following both primary and reinfection. Although in the latter case, it is likely to be more transient and of a lower level One solution for the differentiation of primary from reinfection could be the measurement of the antigen-binding avidity of specific IgG. The avidity of IgG is low after primary antigenic challenge but matures slowly within weeks and months.
Methods
Semi-quantitative test ;- This is based on the recognition of a characteristic pattern in the radial haemolysis test. The zone of haemolysis was assessed; Haemolytic zones with "soft" diffuse outer margins are produced by antibodies of low avidity. Haemolytic zones with a discrete outer margins (designated "ordinary" ) are produced by antibodies of high avidity. Zones that were neither diffuse nor discrete are classified as equivocal.
Quantitative test "Avidity ELISA" ;-
Elution principle - in this test a mild protein denaturing agent such as urea or diethylamine (DEA), is added to the antibody - antigen mixture. Antibodies of low avidity are more likely to dissociate from the antigen-antibody complexes than those of higher avidity. The rest of the test is as for a normal ELISA. The results obtained in the presence and absence of the denaturing agent is compared and a ratio is derived. Low avidity antibodies will have a much higher ratio than high avidity antibodies.
The sera to be tested is diluted at various concentrations in the presence and absence of DEA and assayed for IgG1 and IgG3. The optical densities obtained were plotted. The highest OD (V) was noted and halved (V/2) and the distance between the OD curves at V/2 was measured as the DEA shift value.
Hedman et al (1989) measured the IgG avidity from 64 sera. According to their avidity IgG ELISA, 29 had low avidity Ab, 29 had high avidity Ab and 6 were borderline. Comparison with known clinical records showed that all patients with low avidity Ab had recent primary infection. Those with high avidity had previously been immune. Amongst this group were 4 that had originated from confirmed reinfections. Of the equivocal sera, 5 out of 6 were obtained within 2 months of primary infection.
Thomas and Morgan-Capner (1988) measured the avidities of Rubella specific IgG1 and IgG3. IgG3 is rarely demonstrated in reinfection. They tested sera from 24 patients who were immunized or infected in the distant past, 66 who recent rubella primary infection, 11 from those with symptomatic reinfection and 64 from those with asymptomatic reinfection. For IgG1 the DEA shift value was< 0.6 for cases of rubella in the distant past, compared with 0.8 for the first month after primary infection. The maximum DEA shift value for the sera from cases of reinfection was 0.65. No serum from cases of rubella in the distant past contained sufficient specific IgG3 to estimate avidity. The sera collected within one month of the onset of rubella gave DEA shift values of 0.7 compared to sera from reinfection. In general, the elution-principle test is more sensitive for past infection but less sensitive for recent infection. Whereas the dilution-principle test is more sensitive for recent infection.
10. Molecular Techniques
Molecular biology techniques for the direct detection of viral genomes in the specimen will play an increasingly important role in the clinical virology laboratory in the 21st century. Molecular techniques can be divided into two categories: those that do not involve amplification i.e. hybridization with nucleic acid probes, and those that involve amplification e.g. PCR, LCR, NASBA etc.
Nucleic Acid Probes
Nucleic acid probes are segments of DNA or RNA that have been labeled with enzymes, antigenic substrates, chemiluminescent moeities, or radioisotopes. They can bind with high specificity to complementary sequences of nucleic acid. Probes can be directed to either DNA or RNA targets and can be from 20 to thousands of bases long. The presence and the quantity of hybrids after hybridization is determined by the detection of the label. Probes are usually synthesized by one of the following three methods.
1. Oligonucleotide probes - these are usually less than 50 bases long and are synthesized chemically
2. PCR - will produce probes from 50 to several hundred bases long
3. Cloning - will produce probes several thousand bases long e.g. probe for complete HBV genome
Altering the stringency of the reaction will alter the sensitivity and specificity of the hybridization. Stringency relates to the number of mismatched base pairs that can be tolerated when two nucleic acid molecules come together to form a double stranded molecule. Stringency is affected by several variables, including the temperature, salt concentration, and the pH of the hybridization reaction. High stringency is achieved by using buffers of low salt concentration or by conducting the hybridization reaction and stringency washes at higher temperatures. The higher the stringency of reaction, the less likely it is for mismatched base pairs to stay together.
The hybridization reaction may be carried out completely in solution phase whereby both the target nucleic acid and the probe are free to interact in the reaction mixture. Solution hybridization has the advantages of being rapid to the carry and carry a higher sensitivity than solid phase hybridization. This is the approach taken by Abbott with their quantitative HVB-DNA assay. Nucleic acid bound to a solid surface are still available to participate in hybridization reactions. However, the sensitivity tends to be lower than that of liquid hybridization. However, this technique greatly facilitates the handling of multiple samples. The dot-blot and sandwich hybridization assays are commonly used in this respect. In situ hybridization assays, in which whole cells or tissue sections are put through the hybridization process has become an important research tool.
Despite having been around for many years, hybridization
assays are still not in common use in the clinical virology
laboratory. The main reason is that its sensitivity is not
usually higher than far simpler conventional virological
techniques such as cell culture and viral antigen detection.
Polymerase Chain Reaction
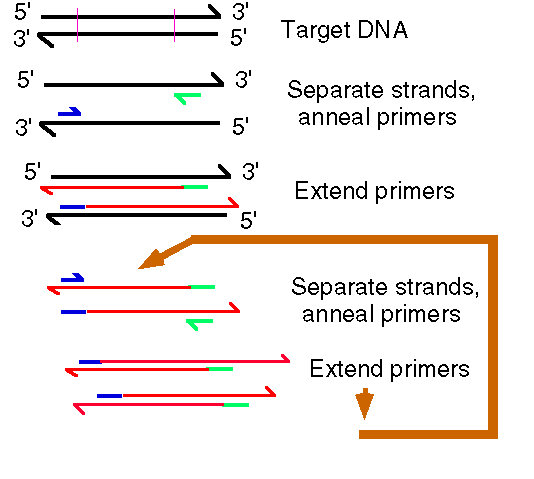
PCR allows the in vitro amplification of specific target DNA sequences by a factor of 106 and is thus an extremely sensitive technique. It is based on an enzymatic reaction involving the use of synthetic oligonucleotides flanking the target nucleic sequence of interest. These oligonucleotides act as primers for the thermostable Taq polymerase. Repeated cycles (usually 25 to 40) of denaturation of the template DNA (at 94oC), annealing of primers to their complementary sequences (50oC), and primer extension (70oC) result in the exponential production of the specific target fragment. Further sensitivity and specificity may be obtained by the nested PCR technique, whereby the DNA is amplified in two steps. In the first step, an initial pair of primers is used to generate a long sequence that contain the target DNA sequence. A small amount of this product is used in a second round of amplification, which employs primers to the final target DNA.
Schematic of Polymerase Chain Reaction
Detection of DNA sequence product of the PCR assay may be performed in several ways. The least sensitive and specific method is to size fractionate the reaction product on an agarose or acrylamide gel and stain the DNA with ethidium bromide. A more sensitive technique involves the attachment of DNA to a membrane through dot or slot-blot techniques followed by hybridization with a labelled homologous oligonucleotide probe. Alternatively, the PCR product may be probed directly by liquid oligomeric hybridization. However, these techniques provides no information on size of the amplified product and thus could not exclude the possibility that the product originated from a region of the human genome which exhibits homology with the target CMV sequence. The most sensitive and specific detection methods result from combining the size information of gel electrophoresis with the improved sensitivity and specificity of hybridization techniques. This may be achieved by gel electrophoresis followed by Southern transfer and hybridization, or through liquid oligomeric hybridization followed by gel electrophoresis.
Advantages of PCR:
Extremely high sensitivity, may detect down to one viral genome per sample volume
Easy to set up
Fast turnaround time
Disadvantages of PCR
Extremely liable to contamination
High degree of operator skill required
Not easy to quantitate results
A positive result may be difficult to interpret, especially with latent viruses such as CMV, where any seropositive person will have virus present in their blood irrespective whether they have disease or not.
The first three problems are being addressed by the arrival of commercial closed systems such as the Roche Cobas Amplicor which requires minimum handling. The use of synthetic internal competitive targets in these commercial assays has facilitated the quantification of results. Otherwise, the same problems remain with in-house PCR assays. The problem with contamination is particularly acute with nested PCR assays and they should be avoided in the clinical setting as far as possible. The sensitivity would normally be sufficient if one use a single PCR reaction followed by hybridization with a specific oligonucleotide probe. This is the approach taken by commercial assays. The fourth problem is more difficult to resolve but it is generally found that patients with active CMV disease has a much higher viral load than those who do not. Therefore, it is simply a case of finding the appropriate cut-off.
Real time quantitative PCR
In real-time PCR, the presence of the PCR product is monitored during the PCR process, not at its end. Real-time PCR can be used to quantify the PCR product. There are two common methods used in the detection of PCR product: non-specific fluorescent dyes and specific DNA probes. The fluorescent dye intercalate with any double-stranded DNA present. Whereas the oligonucleotide DNA probes are labeled with a fluorescent reporter which permits detection only after hybridization of the probe with its target sequence e.g. the Taqman system. Real-time quantitative PCR is being increasingly used for viral diagnosis in routine laboratories.
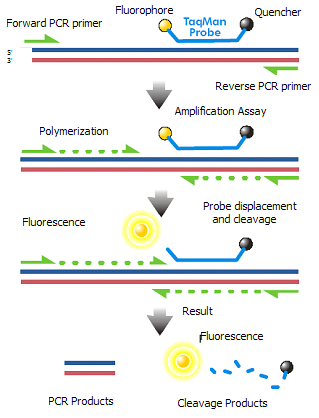
Other Amplification Techniques
Following the heels of PCR, a number of alternative in-vitro amplification techniques have been developed, of which some are now available commercially. Examples of these alternative techniques include ligase chain reaction (LCR), nucleic acid sequence based amplification/isothermal amplification (NASBA), strand displacement amplification, Qb Replicase method, and branched DNA probes. Of these techniques, LCR, NASBA and branched DNA are now available commercially in an automated or semi-automated format. A NASBA assay is available for the quantification of HIV-RNA (Organon), and an LCR assay is available for the detection of chlamydia (Abbott). Branched DNA assays are available for the detection of quantification of HIV-RNA, HBV-DNA, and HCV-RNA (Chiron).
With the exception of the branched DNA probe, all these techniques involve exponential amplification of either the target nuclei acid or the probe. Therefore, they are all as susceptible to contamination as PCR. The branched DNA system is really an intermediate between classical hybridization techniques and the newer in-vitro amplification techniques. It is not as sensitive as those techniques which involve exponential amplification but is considerably more sensitive than the classical hybridization techniques. Below is a brief summary of the features of the different amplification methods available.
![]()
![]()
![]()